はじめに
RDKitには化合物をフラグメントする手法がいくつか用意されています。
その中の一つにBRICSと呼ばれるアルゴリズムがあり、フラグメント化だけでなく
得られたフラグメントから化合物ライブラリの構築が可能です。
(主な使い方については化学の新しいカタチさんの以下の記事をご覧ください。)
上記の通りBRICSモジュールには化学構造生成機能が含まれていますが、これを用いずに
フラグメントの構造修飾・変換を行いたい場合があるかと思います。
例としてA-B型分子、A-B-C型分子の生成をおこなっている記事を紹介します。
上の2つの記事ではフラグメントから分子を作成する際にやや複雑な操作をしていたため、
今回は同様の分子構造生成を行う関数を簡略化してみました。
環境
- Windows 10
- python 3.8.8
- RDKit 2021.03.1
下準備
今回もAsinex社のビルディングブロックを題材として扱います。
まずは以前の記事中で作成したpickleファイルを読み込みます。
import numpy as np
import pandas as pd
from rdkit import Chem
from rdkit.Chem import AllChem, Draw, Descriptors, BRICS
import re
# パッケージの読み込み
df = pd.read_pickle("Asinex_Building_Blocks.pkl")
# Asinex_Building_Blocks.pklを読み込み、得られたデータフレームをdfと定義した
フラグメントの準備
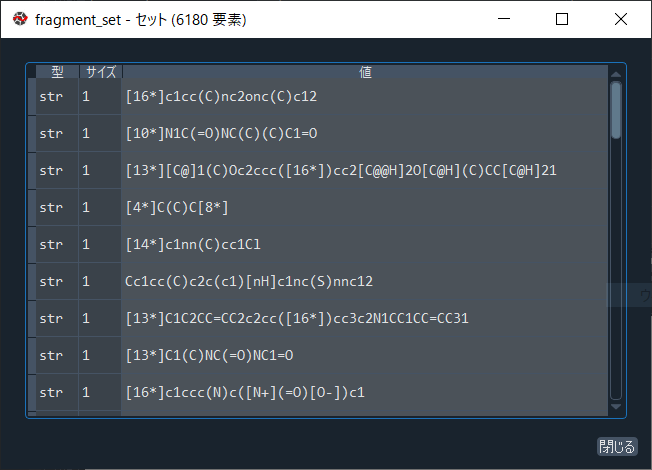
まずは読み込んだ22,525分子をBRICS.BRICSDecompose関数によって分解します。
この分子群からは6,180個のフラグメントが得られます。
(PC環境にもよりますが、この分解作業には約1分の時間を要します)
なお、生成するフラグメントの結合切断箇所にはダミーアトムが挿入されており、
ダミーアトムには結合切断のパターンに応じて固有の番号が振られています。
fragment_set = set()
# 空の集合を用意し、fragment_setと命名した。
for mol in df['ROMol']:
fragment = BRICS.BRICSDecompose(mol)
fragment_set = fragment_set | fragment
# df['ROMol']に格納された分子をフラグメント化し、fragment_setに格納した。
続いてダミーアトム(結合可能部位)が1つのフラグメントをピックアップします。
この際、ダミーアトムに振られた番号を正規表現で置換し、統一して扱えるようにします。
また、複雑な構造のフラグメント(分子量300以上)は削除します。
frag_1dummy = [Chem.MolFromSmiles(re.sub('[0-9]*\*', '100*', x))
for x in fragment_set if x.count('*') == 1]
# ダミーアトムが1つのものをピックアップするとともに、ダミーアトムを[100*]へと置換した。
# ピックアップしたフラグメントをfrag_1dummyに格納した。
frag_1dummy = [x for x in frag_1dummy if Descriptors.MolWt(x) <= 300]
# 分子量が300以下のフラグメントのみをピックアップした。
BRICSで生じるダミーアトムは[1*]から[16*]の16種類あります。
念のためそれらと被らないように今回ダミーアトムは[100*]と置換しました。
上の操作は以下のように一気に書くことも可能です。
frag_1dummy = [Chem.MolFromSmiles(re.sub('[0-9]*\*', '100*', x)) for x in fragment_set
if (Descriptors.MolWt(Chem.MolFromSmiles(x)) <= 300 and x.count('*') == 1)]
同様にダミーアトムが2つのフラグメントも用意しましょう。
frag_2dummy = [Chem.MolFromSmiles(re.sub('[0-9]*\*', '100*', x)) for x in fragment_set
if (Descriptors.MolWt(Chem.MolFromSmiles(x)) <= 300 and x.count('*') == 2)]
# ダミーアトムが2つかつ分子量が300以下のフラグメントをピックアップし、ダミーアトムを[100*]に置換。
# 得られたフラグメントをfrag_2dummyに格納した。
フラグメントを結合して分子を生成する関数の作成
まずはダミーアトムが1つのフラグメント2つを結合し、A-B型の分子を生成する関数を作成します。
def structure_generator_AB_type (frag_1, frag_2):
rxn_AB = AllChem.ReactionFromSmarts("[*:1][100*].[*:2][100*]>>[*:1][*:2]")
return rxn_AB.RunReactants([frag_1, frag_2])[0][0]
同様に、2つのダミーアトムを有する主骨格に対してダミーアトム1つの側鎖2つを結合し、
A-B-C型の分子を生成する関数を作成します。
def structure_generator_ABC_type (core, side_1, side_2):
rxn_ABC = AllChem.ReactionFromSmarts("([*:1][100*].[*:2][100*]).[*:3][100*].[*:4][100*]>>([*:1][*:3].[*:2][*:4])")
return rxn_ABC.RunReactants([core, side_1, side_2])[0][0]
テスト
先に用意したフラグメントからランダムにピックアップし、上記の自作関数の機能を確かめます。
frag_mol_1 = frag_1dummy[np.random.randint(len(frag_1dummy))]
frag_mol_2 = frag_1dummy[np.random.randint(len(frag_1dummy))]
# ダミーアトムが1つのフラグメントをランダムに呼び出し、frag_mol_1およびfrag_mol_2と命名した。
main_mol = frag_2dummy[np.random.randint(len(frag_2dummy))]
# ダミーアトムが2つのフラグメントをランダムに呼び出し、main_molと命名した。
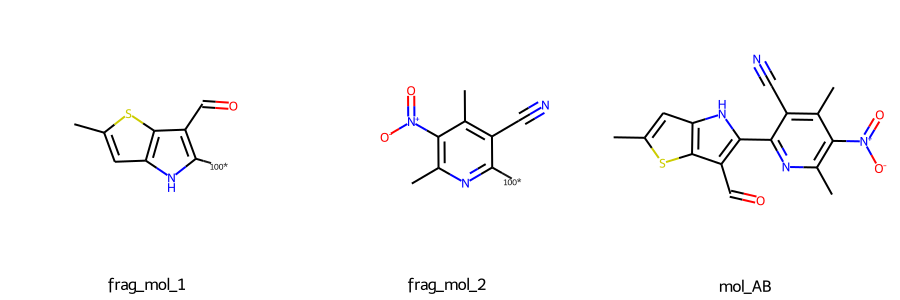
まずはA-B型の分子について。
mol_AB = structure_generator_AB_type(frag_mol_1, frag_mol_2)
Draw.MolsToGridImage([frag_mol_1, frag_mol_2, mol_AB], subImgSize = (300,300),
legends = ["frag_mol_1", "frag_mol_2", "mol_AB"])
続いてA-B-C型の分子について。
mol_ABC = structure_generator_ABC_type(main_mol, frag_mol_1, frag_mol_2)
Draw.MolsToGridImage([frag_mol_1, frag_mol_2, main_mol, mol_ABC], subImgSize = (300,300),
legends = ["frag_mol_1", "frag_mol_2", "main_mol", "mol_ABC"],
molsPerRow = 4)
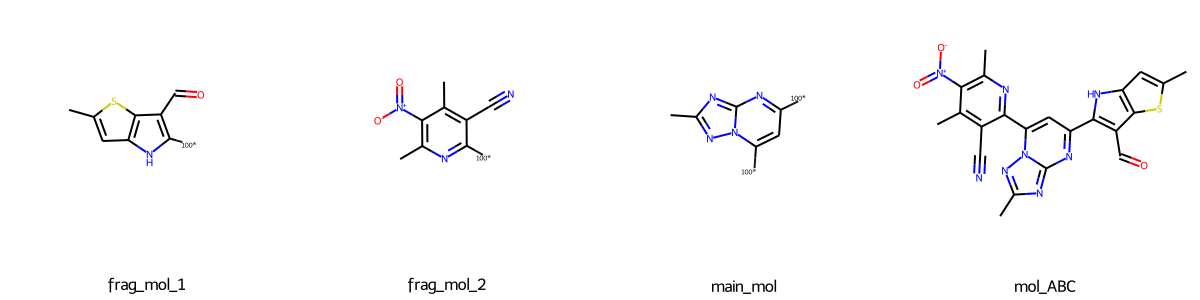
どちらも問題なく構造生成ができています。
冒頭で紹介した2つの記事より関数を簡略化することに成功しました。
おわりに
今回はA-B型およびA-B-C型の分子構造をBRICSフラグメントから生成する関数を作成いたしました。
既存の手法では化合物の隣接行列等を利用して分子生成をおこなっているようでしたが、
ダミーアトムの正規表現置換による統一と化学反応モジュールの使用で関数を簡略化できました。
今回紹介した手法を用いればA-B型、A-B-C型以外にも多様な分子構造を生成することが可能です。
フラグメントのピックアップおよび構造生成の反応式を変更するだけなので、是非試してみてください。
参考
- RDKitでBRICSを用いた仮想ライブラリーの構築
- RDkitを用いた分子操作(分子のフラグメント化)
- BRICSBuildによる分子構造生成
- RDkitを用いた分子構造生成1(A-B型)
- RDkitを用いた分子構造生成2(A-B-C型)
これまでの記事のシリーズ
RDKit入門①:まずは環境構築
RDKit入門②:1分子の読み込みと部分構造検索
RDKit入門③:複数分子の読み込み (前編)と分子群の描画
RDKit入門④:複数分子の読み込み (後編)とデータフレームの加工
RDKit入門⑤:データフレーム内の分子群に対する部分構造検索
RDKit入門⑥:Morganフィンガープリントの作成とそれを用いたタニモト係数の計算による分子類似性評価
RDKit入門⑦:反応式の取り扱い(前編)
RDKitで化学反応を扱った際の生成物群を整理し図示する方法
RDKit入門⑧:反応式の取り扱い(後編)
RDKitで化学反応を繰り返しつつ分子の重複数をカウント・記載する
RDKit入門⑨:データフレーム内の分子群に対する化学反応の適用とExcelファイルへの出力
RDKit入門⑩:データフレーム内の分子群に対する完全一致検索
指定した回数だけ化学反応を実施する関数の作成