はじめに
BRICSによって生成したフラグメントを組み合わせることで新しい分子構造を生成する。(BRICSについて)
前回は、二つのフラグメント(A,B)を結合することで、A-B型の分子を生成したが、今回は、三つのフラグメント(A,B,C)を結合させることで A-B-C型の分子を生成する。
水溶解度データ(金子研究室HPのデータセット)に含まれる1290分子の構造をフラグメント化し構造生成に利用する。
今回のコードは明治大学金子先生の"structure_generator_based_on_r_group"を参考に(コードを改変して)作成しました。(金子先生ありがとうございます)
前準備
モジュールのインストール
conda install -c conda-forge rdkit
コード
モジュールのインポート
import numpy as np
import itertools
from rdkit import rdBase, Chem
from rdkit.Chem import AllChem, Draw, BRICS, Descriptors
from rdkit.ML.Descriptors import MoleculeDescriptors
フラグメントの準備
# データの準備
suppl = Chem.SDMolSupplier('logSdataset1290_2d.sdf')
mols_list = [mol for mol in suppl if mol is not None]
# 1290分子を分解してフラグメントを準備する
fragment_set = set()
for mol in mols_list:
fragment = BRICS.BRICSDecompose(mol)
fragment_set = fragment_set | fragment
fragment_smiles = list(fragment_set)
# フラグメントのリストから、ダミーアトムの数が一つ、二つのものを抽出する。
frag_1dummy_s = [smiles for smiles in fragment_smiles if smiles.count('*')==1]
frag_2dummy_s = [smiles for smiles in fragment_smiles if smiles.count('*')==2]
frag_1dummy = np.array([Chem.MolFromSmiles(smiles) for smiles in frag_1dummy_s])
frag_2dummy = np.array([Chem.MolFromSmiles(smiles) for smiles in frag_2dummy_s])
# 複雑なフラグメント(分子量300以上)は削除する
descriptor_calc = MoleculeDescriptors.MolecularDescriptorCalculator(['MolWt'])
MolWt_list1 = np.array([descriptor_calc.CalcDescriptors(mol)[0] for mol in frag_1dummy])
frag_1dummy = frag_1dummy[np.where(MolWt_list1<=300)]
descriptor_calc = MoleculeDescriptors.MolecularDescriptorCalculator(['MolWt'])
MolWt_list2 = np.array([descriptor_calc.CalcDescriptors(mol)[0] for mol in frag_2dummy])
frag_2dummy = frag_2dummy[np.where(MolWt_list2<=300)]
print('number of fragment:',len(frag_1dummy))
# >>> number of fragment: 333
print('number of fragment:',len(frag_2dummy))
# >>> number of fragment: 130
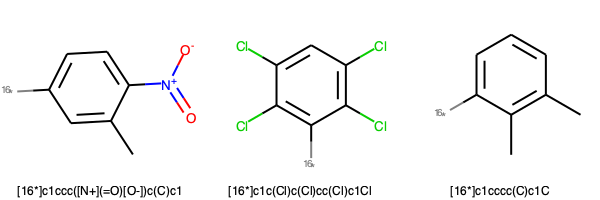
ダミーアトム(*)が一つのフラグメント
img = Draw.MolsToGridImage(frag_1dummy[:3], molsPerRow=3,legends=frag_1dummy_s[:3])
img.save('frag_1dummy.png')
img
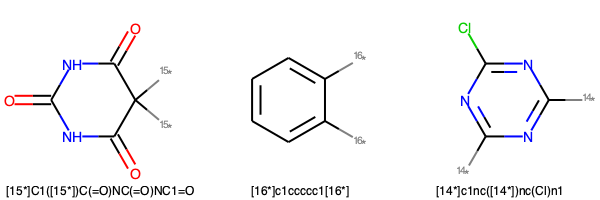
ダミーアトム(*)が二つのフラグメント
img = Draw.MolsToGridImage(frag_2dummy[:3], molsPerRow=3,legends=frag_2dummy_s[:3])
img.save('frag_2dummy.png')
img
構造生成する関数を作成する
フラグメントの一つを主骨格(main_mol,A)とみなしてそこにフラグメント(fragment1,2:B,C)を結合するテイ。
def structure_generator(main_mol, fragment1, fragment2, r_position=1):
"""
parameters
----------
main_mol : RDkit Mol object
主骨格(A)の構造(フラグメント)
fragment1 : RDkit Mol object
* の数が二つのフラグメント(Bにあたる)
fragment2 : RDkit Mol object
* の数が一つのフラグメント(Bにあたる)
r_position : int (1 or 2)
fragment1 中の * のどちらに、main_mol, fragment1を結合させるか。
Returns
----------
generated_molecule : RDkit Mol object
各フラグメントを結合させて生成したA-B-C型の分子
"""
bond_list = [Chem.rdchem.BondType.UNSPECIFIED, Chem.rdchem.BondType.SINGLE, Chem.rdchem.BondType.DOUBLE,
Chem.rdchem.BondType.TRIPLE, Chem.rdchem.BondType.QUADRUPLE, Chem.rdchem.BondType.QUINTUPLE,
Chem.rdchem.BondType.HEXTUPLE, Chem.rdchem.BondType.ONEANDAHALF, Chem.rdchem.BondType.TWOANDAHALF,
Chem.rdchem.BondType.THREEANDAHALF, Chem.rdchem.BondType.FOURANDAHALF, Chem.rdchem.BondType.FIVEANDAHALF,
Chem.rdchem.BondType.AROMATIC, Chem.rdchem.BondType.IONIC, Chem.rdchem.BondType.HYDROGEN,
Chem.rdchem.BondType.THREECENTER, Chem.rdchem.BondType.DATIVEONE, Chem.rdchem.BondType.DATIVE,
Chem.rdchem.BondType.DATIVEL, Chem.rdchem.BondType.DATIVER, Chem.rdchem.BondType.OTHER,
Chem.rdchem.BondType.ZERO]
# make adjacency matrix and get atoms for main molecule
level1_adjacency_matrix = Chem.rdmolops.GetAdjacencyMatrix(fragment1)
for bond in fragment1.GetBonds():
level1_adjacency_matrix[bond.GetBeginAtomIdx(), bond.GetEndAtomIdx()] = bond_list.index(bond.GetBondType())
level1_adjacency_matrix[bond.GetEndAtomIdx(), bond.GetBeginAtomIdx()] = bond_list.index(bond.GetBondType())
level1_atoms = []
for atom in fragment1.GetAtoms():
level1_atoms.append(atom.GetSymbol())
# * の位置を検索
r_index_in_level1_molecule_old = [index for index, atom in enumerate(level1_atoms) if atom == '*']
# matrix,atom の並び替え
for index, r_index in enumerate(r_index_in_level1_molecule_old):
modified_index = r_index - index
atom = level1_atoms.pop(modified_index)
level1_atoms.append(atom)
tmp = level1_adjacency_matrix[:, modified_index:modified_index + 1].copy()
level1_adjacency_matrix = np.delete(level1_adjacency_matrix, modified_index, 1)
level1_adjacency_matrix = np.c_[level1_adjacency_matrix, tmp]
tmp = level1_adjacency_matrix[modified_index:modified_index + 1, :].copy()
level1_adjacency_matrix = np.delete(level1_adjacency_matrix, modified_index, 0)
level1_adjacency_matrix = np.r_[level1_adjacency_matrix, tmp]
r_index_in_level1_molecule_new = [index for index, atom in enumerate(level1_atoms) if atom == '*']
# *がどの原子と結合しているか調べる。
r_bonded_atom_index_in_level1_molecule = []
for number in r_index_in_level1_molecule_new:
r_bonded_atom_index_in_level1_molecule.append(np.where(level1_adjacency_matrix[number, :] != 0)[0][0])
#結合のタイプを調べる
r_bond_number_in_level1_molecule = level1_adjacency_matrix[
r_index_in_level1_molecule_new, r_bonded_atom_index_in_level1_molecule]
# *原子を削除
level1_adjacency_matrix = np.delete(level1_adjacency_matrix, r_index_in_level1_molecule_new, 0)
level1_adjacency_matrix = np.delete(level1_adjacency_matrix, r_index_in_level1_molecule_new, 1)
for i in range(len(r_index_in_level1_molecule_new)):
level1_atoms.remove('*')
level1_size = level1_adjacency_matrix.shape[0]
# level1フラグメントの 2つの* に残りのフラグメントを結合する
generated_molecule_atoms = level1_atoms[:]
generated_adjacency_matrix = level1_adjacency_matrix.copy()
frag_permutations = list(itertools.permutations([main_mol, fragment2]))
fragment_list = frag_permutations[r_position]
for r_number_in_molecule, fragment_molecule in enumerate(fragment_list):
#ここから、主骨格の処理と同じ
fragment_adjacency_matrix = Chem.rdmolops.GetAdjacencyMatrix(fragment_molecule)
for bond in fragment_molecule.GetBonds():
fragment_adjacency_matrix[bond.GetBeginAtomIdx(), bond.GetEndAtomIdx()] = bond_list.index(
bond.GetBondType())
fragment_adjacency_matrix[bond.GetEndAtomIdx(), bond.GetBeginAtomIdx()] = bond_list.index(
bond.GetBondType())
fragment_atoms = []
for atom in fragment_molecule.GetAtoms():
fragment_atoms.append(atom.GetSymbol())
# integrate adjacency matrix
r_index_in_fragment_molecule = fragment_atoms.index('*')
r_bonded_atom_index_in_fragment_molecule = \
np.where(fragment_adjacency_matrix[r_index_in_fragment_molecule, :] != 0)[0][0]
# 後で * を削除するのでそのための調整(たぶん)
if r_bonded_atom_index_in_fragment_molecule > r_index_in_fragment_molecule:
r_bonded_atom_index_in_fragment_molecule -= 1
fragment_atoms.remove('*')
fragment_adjacency_matrix = np.delete(fragment_adjacency_matrix, r_index_in_fragment_molecule, 0)
fragment_adjacency_matrix = np.delete(fragment_adjacency_matrix, r_index_in_fragment_molecule, 1)
#新たに生成する分子用のマトリックス作成
main_size = generated_adjacency_matrix.shape[0]
generated_adjacency_matrix = np.c_[generated_adjacency_matrix, np.zeros(
[generated_adjacency_matrix.shape[0], fragment_adjacency_matrix.shape[0]], dtype='int32')]
generated_adjacency_matrix = np.r_[generated_adjacency_matrix, np.zeros(
[fragment_adjacency_matrix.shape[0], generated_adjacency_matrix.shape[1]], dtype='int32')]
#マトリックスに結合のタイプを記述
generated_adjacency_matrix[r_bonded_atom_index_in_level1_molecule[r_number_in_molecule],
r_bonded_atom_index_in_fragment_molecule + main_size] = \
r_bond_number_in_level1_molecule[r_number_in_molecule]
generated_adjacency_matrix[r_bonded_atom_index_in_fragment_molecule + main_size,
r_bonded_atom_index_in_level1_molecule[r_number_in_molecule]] = \
r_bond_number_in_level1_molecule[r_number_in_molecule]
generated_adjacency_matrix[main_size:, main_size:] = fragment_adjacency_matrix
#フラグメントのマトリックスを入力(マトリックスの右下)
# integrate atoms
generated_molecule_atoms += fragment_atoms
# generate structures
generated_molecule = Chem.RWMol()
atom_index = []
for atom_number in range(len(generated_molecule_atoms)):
atom = Chem.Atom(generated_molecule_atoms[atom_number])
molecular_index = generated_molecule.AddAtom(atom)
atom_index.append(molecular_index)
for index_x, row_vector in enumerate(generated_adjacency_matrix):
for index_y, bond in enumerate(row_vector):
if index_y <= index_x:
continue
if bond == 0:
continue
else:
generated_molecule.AddBond(atom_index[index_x], atom_index[index_y], bond_list[bond])
generated_molecule = generated_molecule.GetMol()
return generated_molecule
テスト
main_mol = frag_1dummy[0]
fragment1 = frag_2dummy[1]
fragment2 = frag_1dummy[1]
mol = structure_generator(main_mol, fragment1, fragment2)
img = Draw.MolsToGridImage([main_mol, fragment1, fragment2, mol],
molsPerRow=4,
legends=['main_mol','fragment1','fragment2','generated_molecule'])
img.save('generated_molecule.png')
img
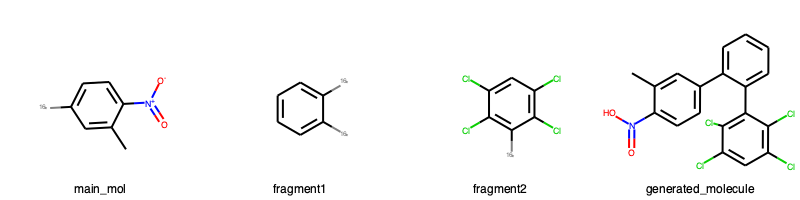
上手く分子を作れた。