はじめに
AlphaFold2時代の構造解析チュートリアルシリーズの連載第3回目(初回, 第2回目).
今回は構造因子$F(hkl)$から初期位相を求める. タイトルに"AlphaFold2時代の”と銘打っているように, AlphaFold2で出力される高精度なモデル構造を利用して初期位相を求める.
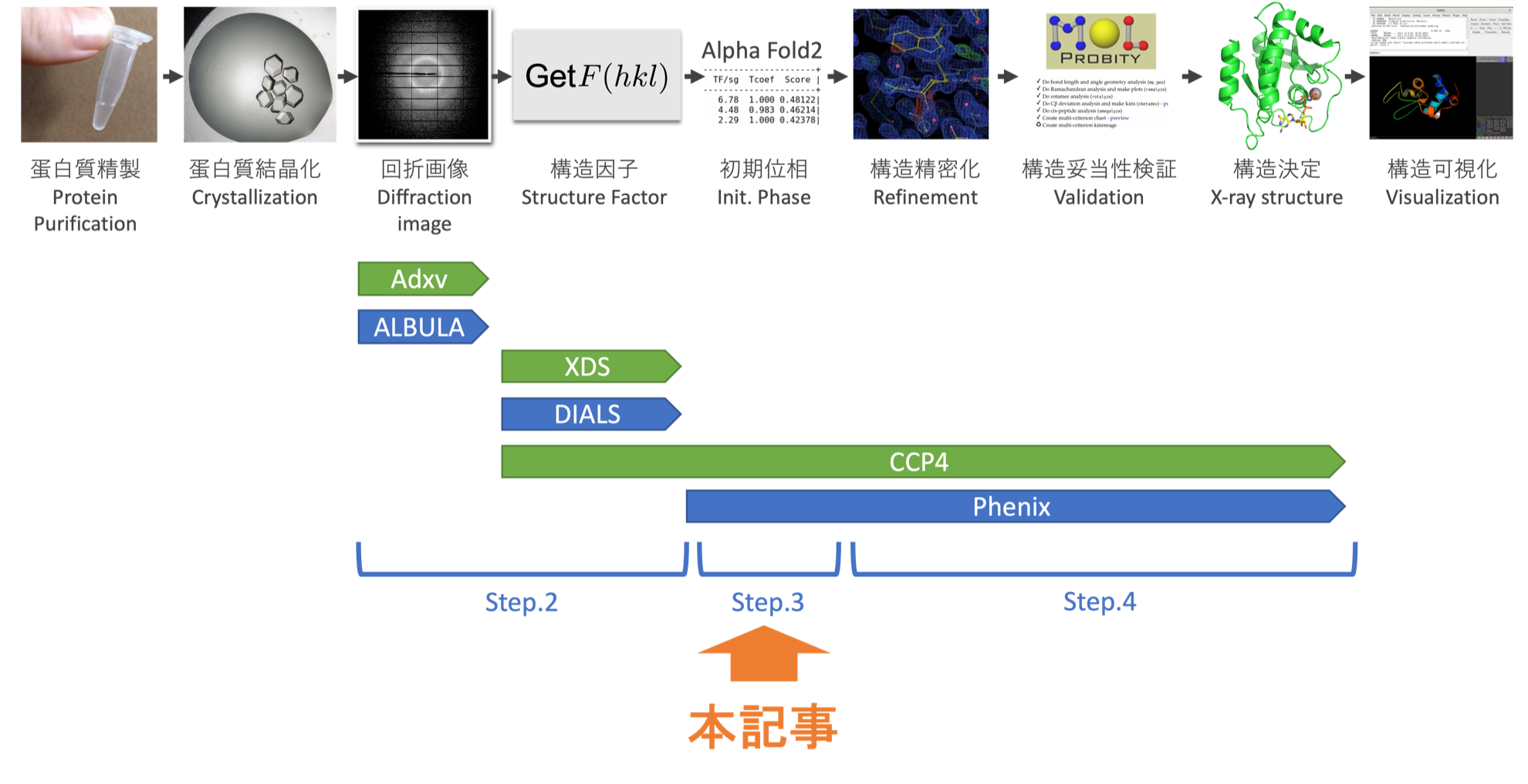
目次
- Step.0 構造決定の流れ
- Step.1 解析ソフトのインストール
- Step.2 回折画像の確認と構造因子の計算
- Step.3 初期位相を求める (本記事)
- Step.4 構造精密化とバリデーション
この記事の対象と目的
タンパク質X線結晶構造法を専門外とする研究者・大学院生・学部生を対象とし, タンパク質X線結晶構造解析について回折画像から構造決定まで一通りできるようになることを目的とする. 前提として, Linux環境でBashコマンドをある程度打てる方を想定している.
本記事では, タンパク質の構造解析のチュートリアルを全4回の連載でお届けする. 今回は第3回目である.
また, 本記事は完全にオープンである. 学校でも研究機関でも企業でも, ぜひこの記事を広めて, 教育に活用していただければ幸いである. しかし, 著作権は放棄していない.
タンパク質のX先結晶構造解析に必要な計算機環境
筆者の経験から, タンパク質のX線結晶構造解析には以下の計算機環境が適していると考えている. コンピューターパワーが必要であるため, 間違ってもRaspberry Piなどでやってはいけない.
- OS
- Ubuntu, CentOSなどのLinux (強く推奨)
- MacOSX
- Windows 10/11 (ネイティブ環境もしくはWSL2上のLinux環境を使用)
- CPU: > 4 core (推奨:> 16 core)
- Main Memory: > 16 GB (推奨: > 32 GB)
- Storage: > 50 GB (ソフトウェアインストールに約 12 GB.データセット 20 - 30 GB/crystal)
- GPU:必須ではない(推奨: GPU搭載)
本StepではStep.1で構築したGoogle Cloud Platform上の仮想マシン(Ubuntu 20.04LTE)で解析を行う.
Githubレポジトリ
本チュートリアルに関連する代表的なファイルはGithubにアップロードしている. 適宜ご参考いただければ幸いである.
Step.3 初期位相を求める
Step.3-0 準備: 初期位相・MR法とは
Step.0で解説したとおり, 位相問題はタンパク質X線結晶構造解析において泣き処である. これはひとえに現在の2次元検出器(例:DECTRIS社のEIGER検出器)では位相$\alpha(hkl)$を直接観測出来ないためである. ではどのように算出するのか. これは非常に奇妙な話に聞こえるかもしれないが 構築したタンパク質のモデル構造(これが得たい)から位相$\alpha(hkl)$を逆算し, 電子雲$\rho(xyz)$を得る. しかし, そのタンパク質のモデル構造するには, そもそも電子雲$\rho(xyz)$が必要なのである1. このように, タンパク質のモデル構造と位相$\alpha(hkl)$は"鶏が先か、卵が先か"問題のようにお互いがお互いに依存する関係であり, タンパク質のモデル構造を得ることを難しくしている.
このままでは目的のタンパク質モデル構造を獲得することはできない. そこで, はじめにモデルをある程度構築できる程度の電子雲$\rho (xyz)$を得るため, 初期の粗い位相$\alpha (hkl)$(初期位相)を求める. その後, 導出した初期位相を用いてタンパク質モデル構造をある程度構築し, それを元に新たな位相$\alpha (hkl)$と新たな電子雲$\rho(xyz)$が計算される. その後は改善された電子雲$\rho(xyz)$を元にモデル構造を改善する. このサイクルを繰り返し, 徐々にタンパク質のモデル構造を精密化してゆく. この工程を 構造精密化(Refinement) という.
さて, 位相問題は低分子のX線結晶構造解析(無機,体有機化合物)の分野ではコンピューターパワーを活かした直接法(Direct method)によって解決できるが, 分子量が大きい(=単位胞が大きい)タンパク質のX線結晶構造解析ではこの方法は使えない. そのため, 偉大な先人は初期位相を求める数々の方法を開発してきた. それは主に①実験的手法(MIR法, MAD法, SAD法など), ②計算的な方法(MR法)に分けられる. ①実験的手法はタンパク質に一定量のCysteineやMethionineなどの硫黄原子(S)が含まれている場合2以外は, 重原子をタンパク質結晶内部に入れ込むなど, 基本的に追加の実験が必要であり, 時間とお金と労力がかかる. 一方, ②計算的な方法では, 類似タンパク質構造を利用した分子置換法(Molecular Replacement法, MR法)が用いられ, これは追加実験の必要はなく, すべて計算機上で実施できるため非常に簡便 である.
筆者の経験になるが, 例えば実験的手法(Ptなどの重原子を用いたSAD法)で1~2ヶ月かかるところ, MR法では5分程度で初期位相の決定が完了する. このように非常に簡便なため, MR法は初期位相を求める最初の一手としてよく選択される. しかし, 類似構造がどうしても見つけられない場合はどうしようもなく3, ①実験的な初期位相決定手法に頼る他なかった.
この状況を一変させたのが, DeepMind社が開発したAlphaFold2である. これまで, MR法に使用する類似構造は配列等の情報を足がかりにProtein Data Bank (PDB)から構造データを入手してきた. しかしながら, タンパク質構造はアミノ酸配列によって千差万別であり, 配列が似ている(30%のアミノ配列相同性があれば類似構造の可能性有りと言われてきた)といっても, 全体構造を変化させるクリティカルなアミノ酸変異の影響で解きたい構造と類似構造が大幅に異なる場合も少なくなく, 構造決定は時に困難であった. ここで登場するのが高精度タンパク質構造予測モデルのAlphaFold2である. PDBから類似構造を見つけ出すこと無く, AlphaFold2が出力するタンパク質モデル構造を, MR法に用いる類似構造とすれば非常に高精度な初期位相を得られると期待される. 実際、Twitter上では構造生物学者の多くから驚きの声が上がっている(例). このように, 今後AlphaFold2 + MR法の利用は初期位相決定法の主流メソッドになると期待され, 本チュートリアルでもそれに則って初期位相を導出してゆく.
Step.3-1 AlphaFold2を用いたモデル構造の準備
本章ではAlphaFold2を用いて, MR法に入力するモデル構造を出力する. しかし, DeepMind社が発表した純正のAlphaFold2は巨大なDataBase(約2.6TB)と高価なGPU (タンパク質配列の長さに比例して大容量VRAMを使う)が必要になり, 気軽に使うにはハードルが高い. よって今回は, Googleが無償で提供しているPython解析実行プラットフォームであるGoogle Colablatory上で作動するAlphaFold2の実装の一つColabFoldで構造を求める. AlphaFold2およびColabFoldの詳細については開発者のお一人である東大森脇先生のQiitaの記事、そしてJSBi Bioinformatics Reviewで発表された総説を参考にしてほしい.
-
ColabFoldのGitHubレポジトリにアクセスし, "AlphaFold2_mmseq2"のリンクからGoogle ColaboratoryのNotebooksを起動する. なお, Google Colaboratoryの使用にはGoogleアカウントは必要なので注意されたい.

2. 基本的な使用法は開いたNotebookに書いてある通りである. query_sequenceにLysozymeの配列を入力する. この際, 配列にスペースを入れないよう気をつける. また, 任意ではあるがjobnameもわかりやすい名前を入力する. その後RuntimeからRun allを選択し, モデル構造の推論を開始する. 処理終了後, 自動的にzipファイルがダウンロードされる. なお, Warningが表示されたらRun anywayを選択する.
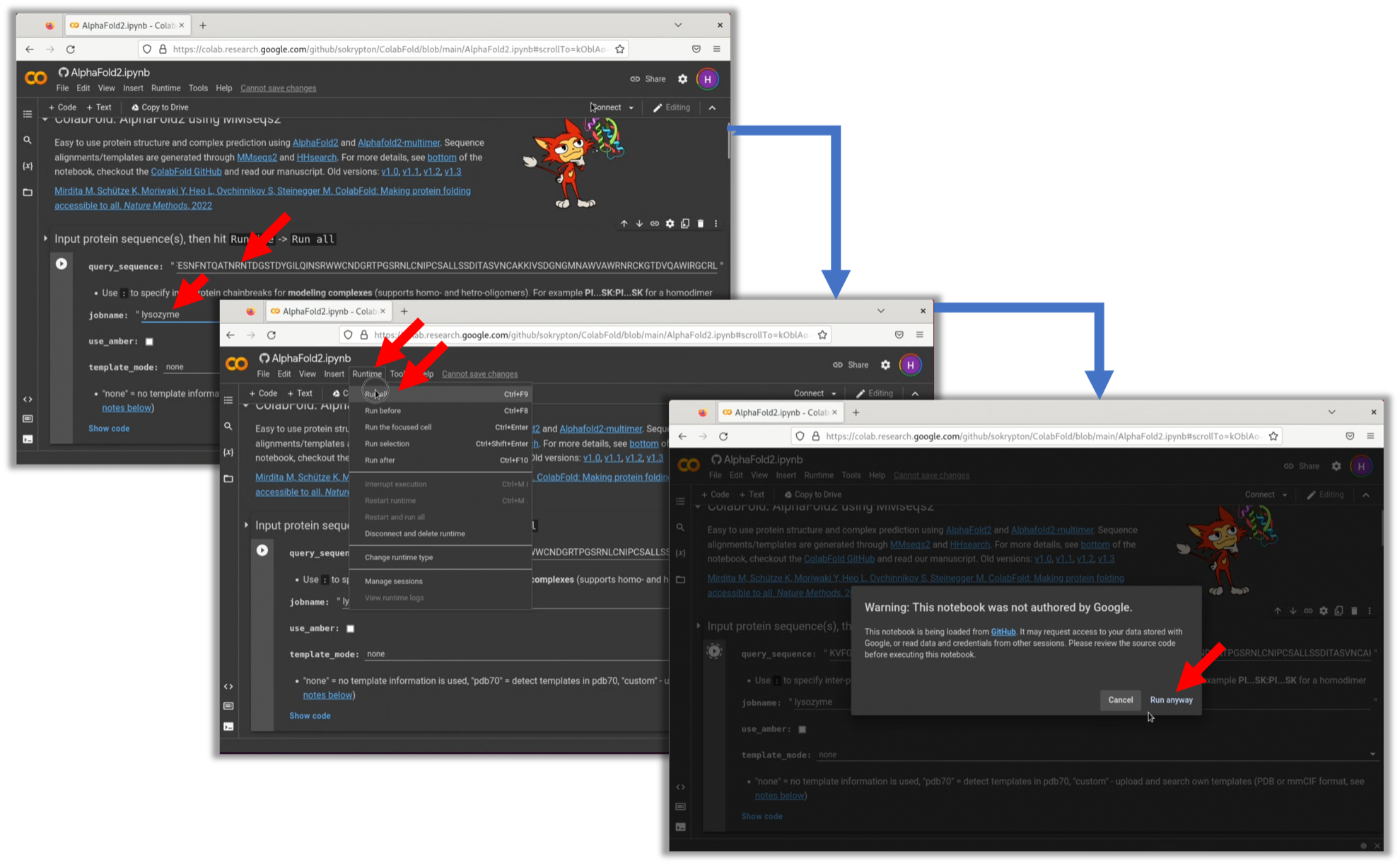
KVFGRCELAAAMKRHGLDNYRGYSLGNWVCAAKFESNFNTQATNRNTDGSTDYGILQINSRWWCNDGRTPGSRNLCNIPCSALLSSDITASVNCAKKIVSDGNGMNAWVAWRNRCKGTDVQAWIRGCRL
筆者でColabFoldで推論したLysozymeのモデル構造をここにアップロードした. 適宜使用していただきたい.
Step.3-2 MR法による初期位相の決定
先に入手したAlphaFold2による推論モデル構造を用いて, 分子置換法(Molecular Replacement法, MR法)を行う.
Step.3-2-1 構造因子F(hkl)(空間群:P41212)を用いたMR法
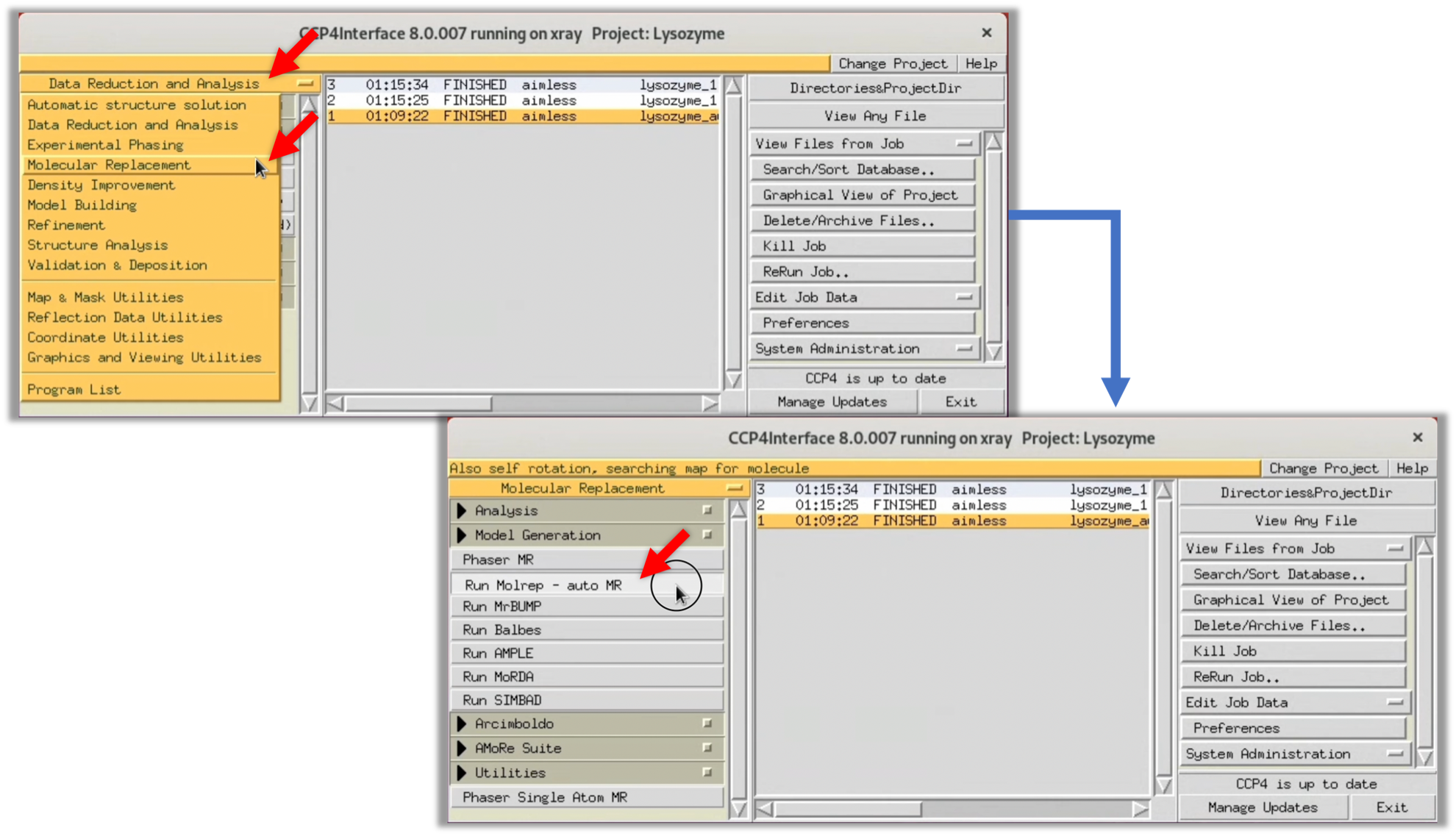
- CCP4iを立ち上げ, 左側のメニューから
Data Reduction and Analysis-->Molecular Replacementを選択し, 分子置換法(Molecular Replacement法, MR法)ソフトMolrepを起動する.
$ ccp4i
2. 下記画像を参考に, 起動したMolrepのWindowに必要な項目を入力する. "Data"にはStep.2で最終的に得られたScaling済みの構造因子$F(hkl)$を指定する. "Model"にはStep.3-1で予測したタンパク質のモデル構造を指定する. "Solution"は自動で入力されるが, 状況によって適当なファイル名を指定する. 入力が終了したら, Run-->Run Nowを選択し, MR法を開始する.
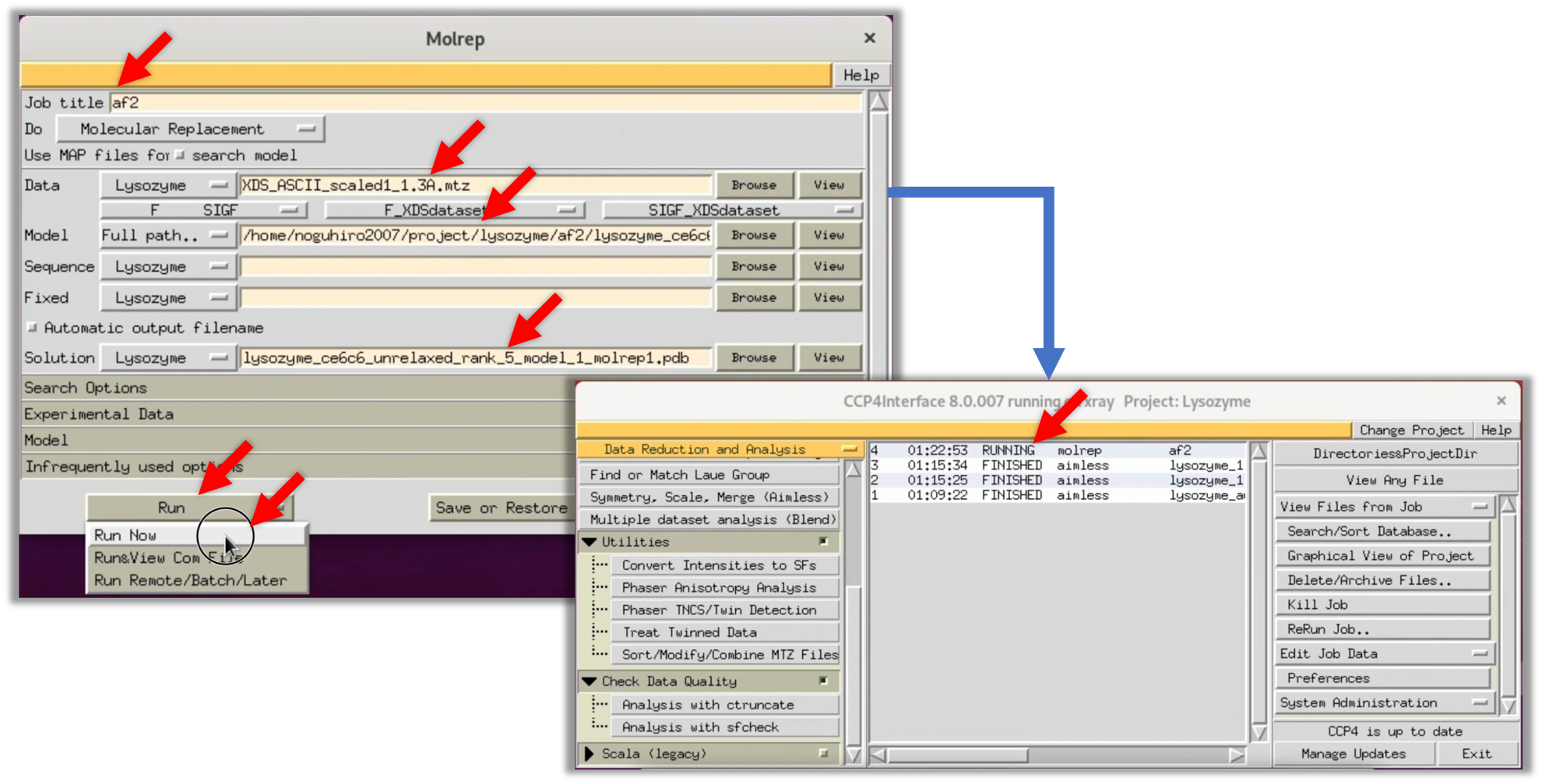
ColabFoldの標準設定では5つの予測モデル構造が出力されるが, MR法にはその中から適当なものを使用する.
3. 処理が終了するとStatusが"FINISHED"に変わる. その項目をダブルクリックしするとGUIの画面でログを見ることができる. そこからLog FileタブをクリックするとMOLREPから出力された実際の元Logを見ることができる. この際, 注目すべきなのは"--- Summary (V2) ---"表におけるTF/sg列とScore列である(赤ハイライトで示す). 一般的に正しい初期位相が求められた際は, TF/sgおよびScoreが他の解に比べて有意に高い.
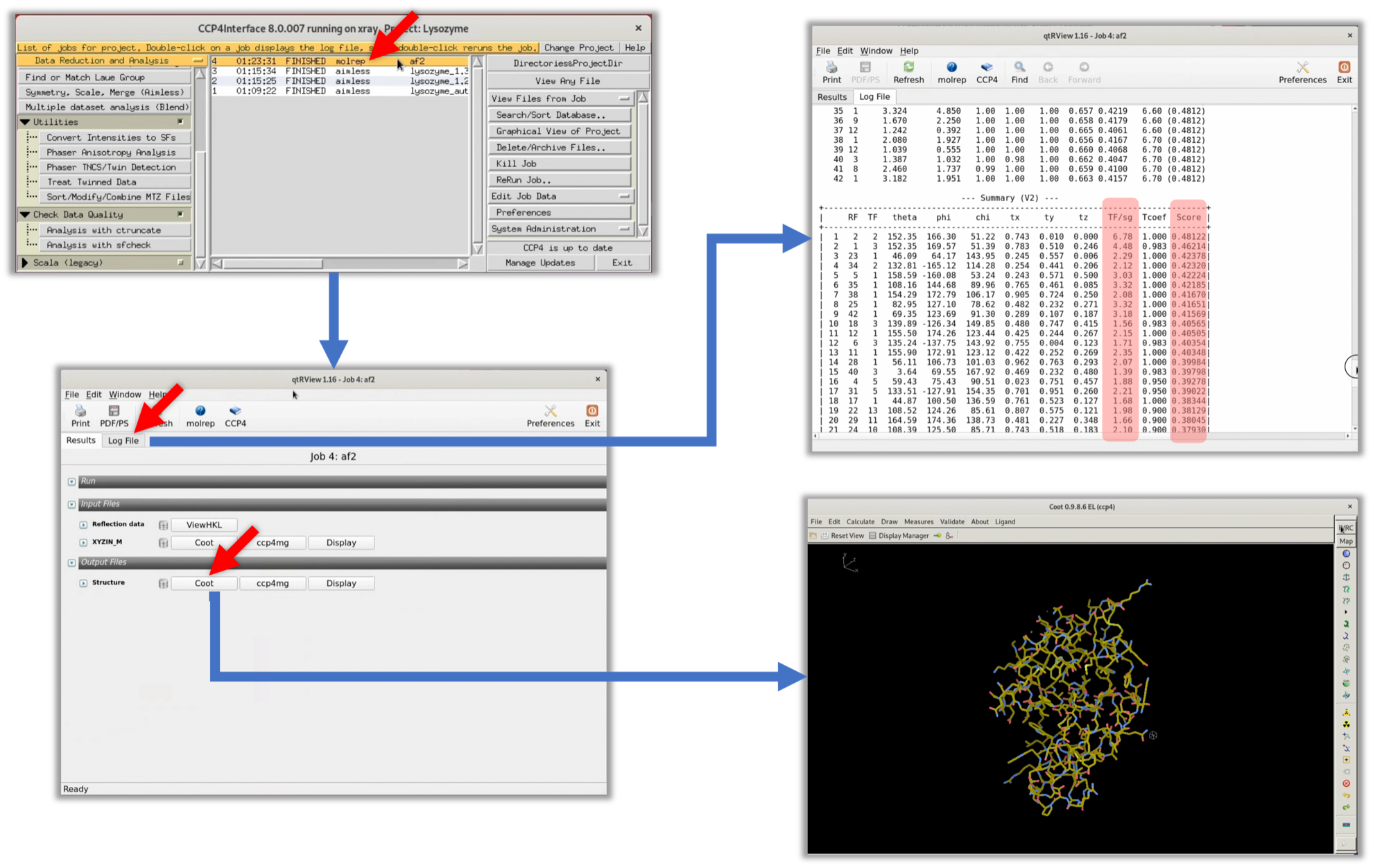
今回のMOLREPのLogにおけるTF/sg列, 及びScoreの値は他の値に比べ優位に高くない. よって, 残念ながら適切な初期位相が導出されていない可能性が高い.
4. MOLREPのLogにおけるTF/sg列及びScoreの値から適切な解が得られた可能性は低いが, 自らの目で電子雲マップ$\rho (xyz)$を確認する. そのためにはRefinementのソフトウェアであるRefmacを用いて, 1回精密化を行って電子雲マップ$\rho (xyz)$を生成する. CCP4iメニューからRefinement-->Run Refmac5を選択し, Refmacのパラメータ入力Windowを表示する.
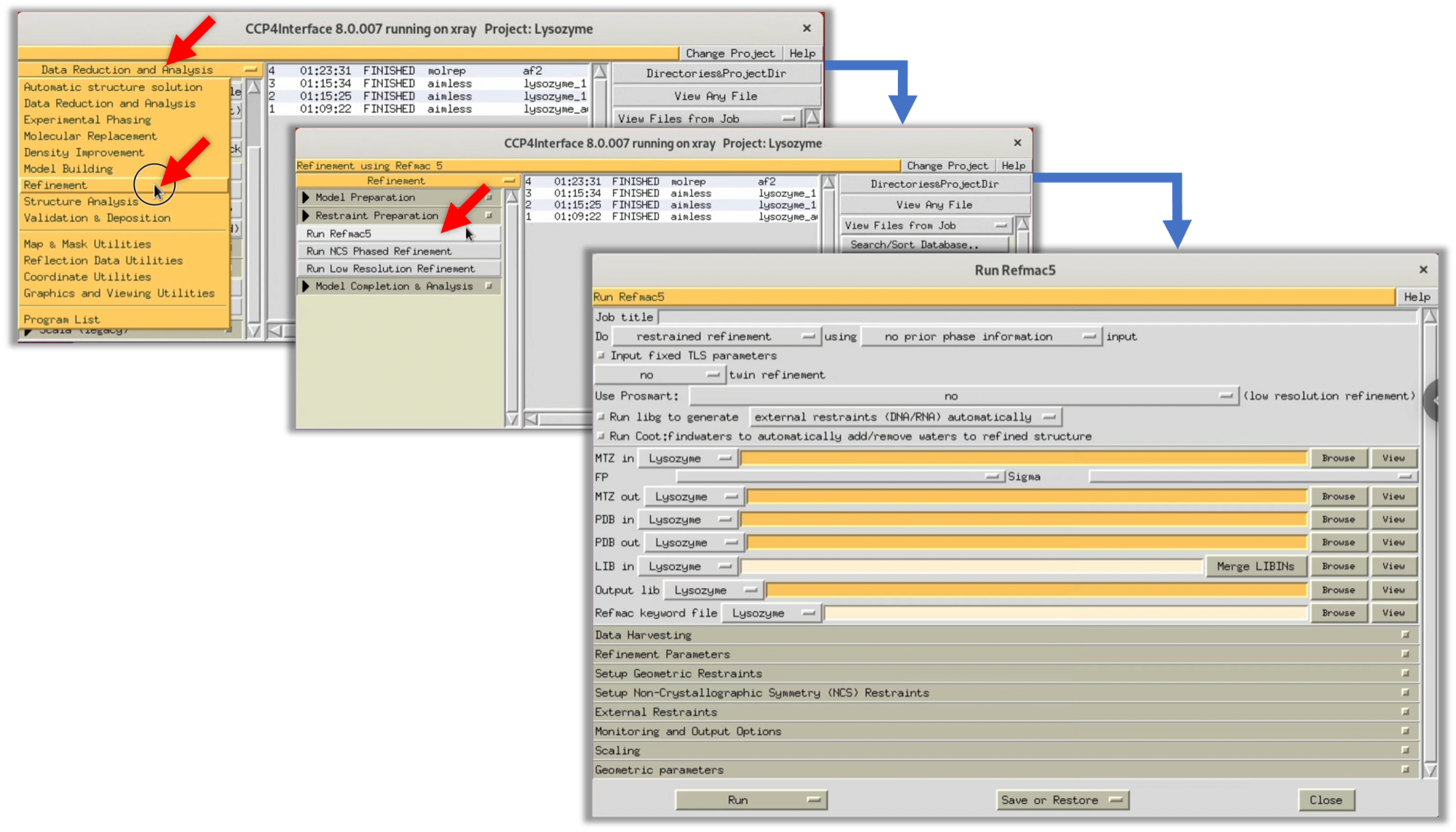
5. 図の赤矢印部分を参考に必要なパラメータを入力し, Run-->Run Nowを選択し, Refmacを開始する.
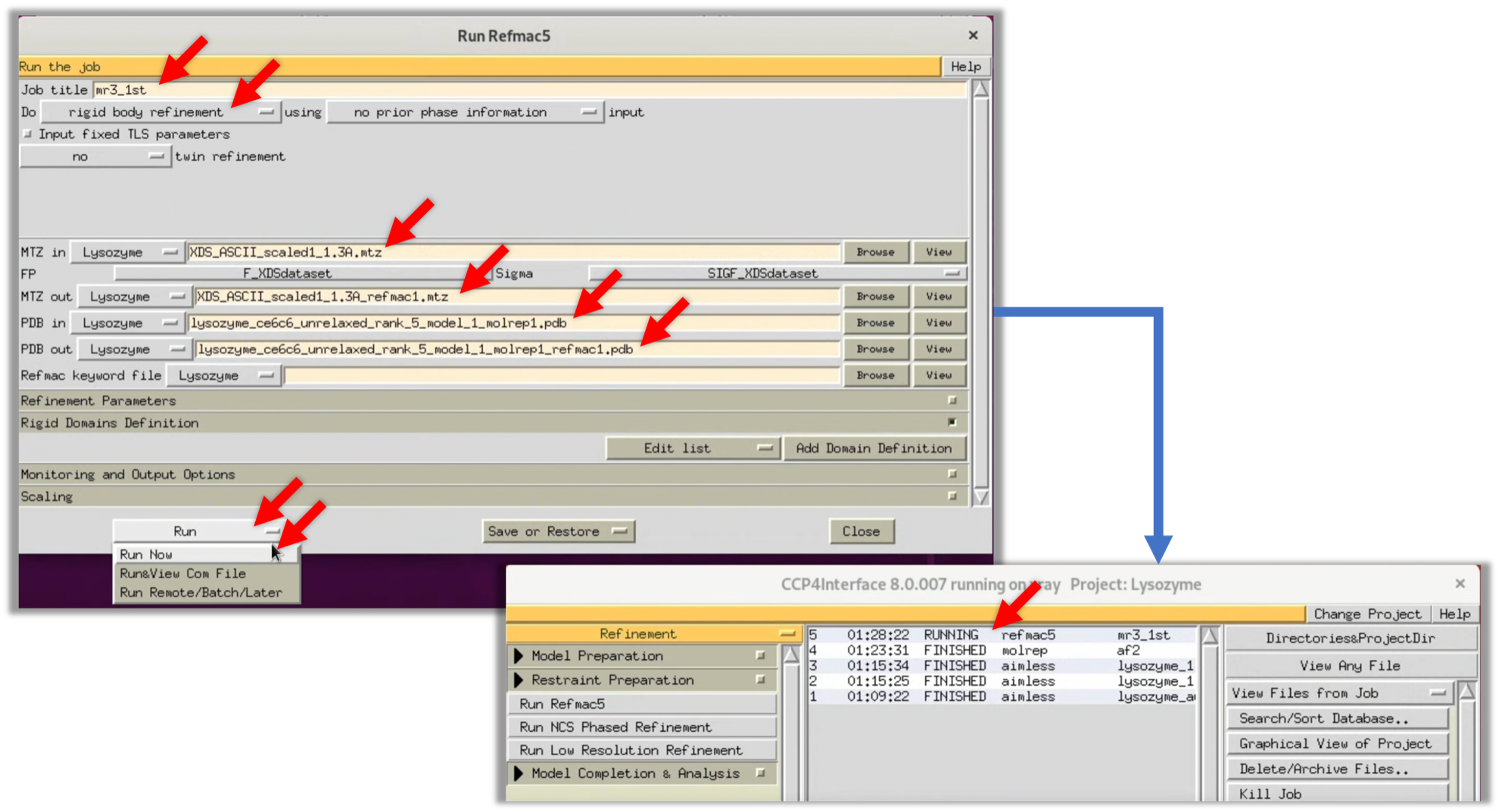
6. Refmacの処理が終了(FINISHED)したら, その項目をダブルクリックし, 表示されたWindowの一番下のCootボタンをクリックし, 本分野でよく使われるタンパク質構造やマップビューワーであるCootを起動する.
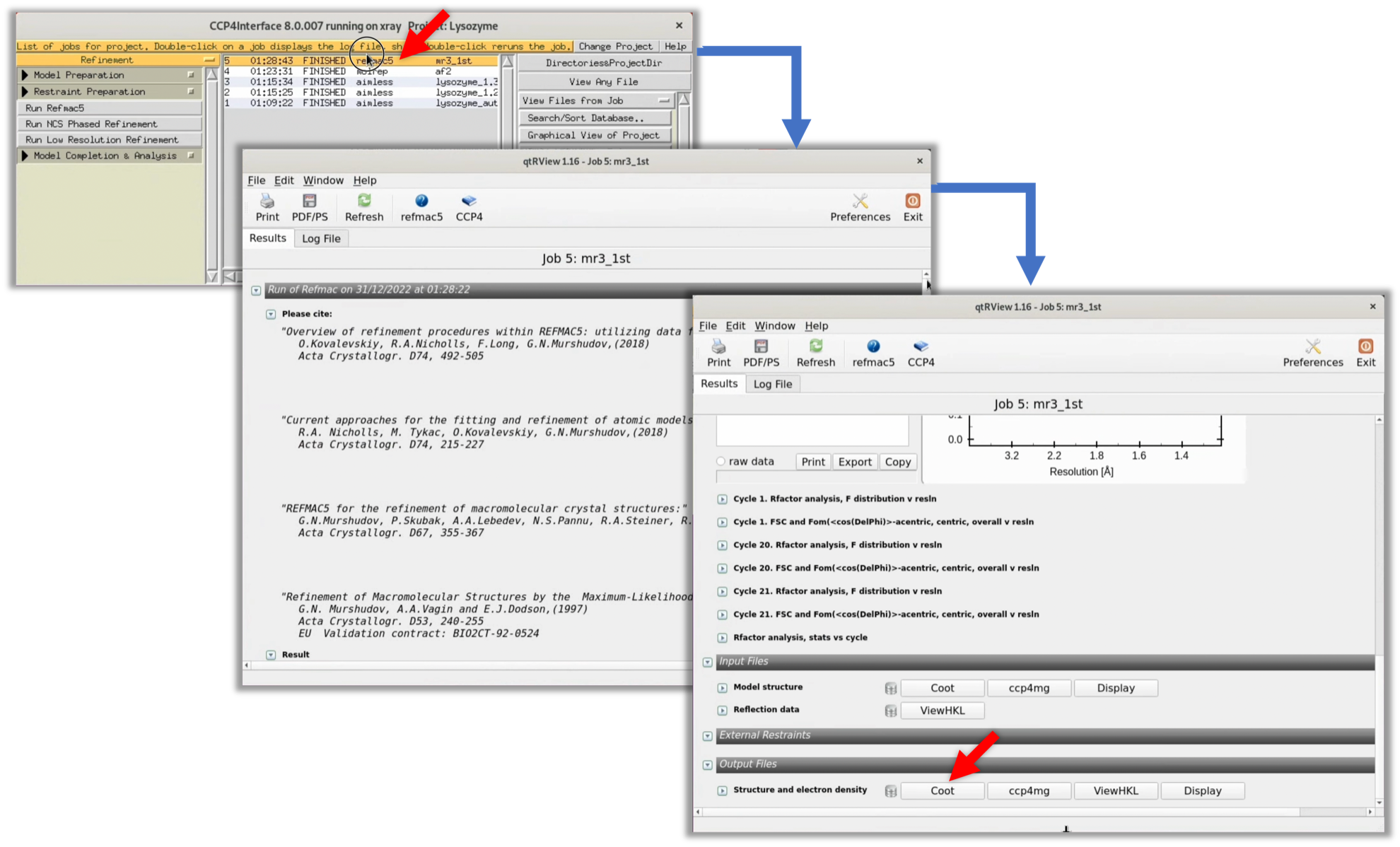
Cootを起動後, 以下の動画を参考に電子雲マップ$\rho (xyz)$を確認する.
特に注目したいのは, タンパク質モデルを全体表示した際, 横のLysozymeの間の溶媒空間と, Lysozymeそのものの電子雲マップ$\rho (xyz)$の濃度差である(動画:2:00 ~). 動画では電子雲マップ$\rho (xyz)$の可視化閾値を変化させても, 明確に両者の区別が難しく電子雲マップ$\rho (xyz)$がランダムに存在する印象がある. この場合, 良質な初期位相が得られていないと判断される.
経験則ではあるが, 一般的にタンパク質モデル本体と周りの溶媒空間の電子雲マップの濃度比が明確であれば, 良質な初期位相が得られたと判断できる.
著者が行った空間群P41212におけるMolrep, Refmacの処理結果をgithubにアップロードした. 適宜参考にしていただきたい.
Step.3-2-2 構造因子F(hkl)(空間群:P41212)でのMR法がうまく行かなかった原因
本チュートリアルのように, Scaling(Step.2-5-2参照)の結果が良好な場合, 同じブラベ格子に属する別の空間群(Space group)が真の空間群である可能性が高い. 上記で処理した構造因子$F(hkl)$は空間群P41212で処理されたものであるが, これを空間群P43212で処理し直し, MolrepでMR法をやり直す.
今回Lysozymeの結晶のブラベ格子はPrimitive Tetragonalであり, その中に属する空間群からP41212がAIMLESSによって予測された. しかし, P41212とP43212は同じらせん軸上の消滅則を有し, 両者をAIMLESSのみで見分けるのは至難の業である. このような場合, 実際に初期位相を求めるまで(場合によっては最終モデル構造を求めるまで)空間群が決定できない場合は珍しくなく, 決定には深い結晶学の知識が必要になる. 一方で, 計算機能力が向上している現代においては, 「Molrepで一番良いスコアが得られた空間群を真の空間群」とする空間群特定戦略も有効だと考えられる.
タンパク質の空間群についてより詳しい解説は, 東京大学大学院 農学生命科学研究科 応用生命工学専攻 伏信研究室の「タンパク質結晶のとりうる空間群の表」記事が大変わかりやすいため, 参考にしていただきたい.
Step.3-2-3 空間群P43212の構造因子F(hkl)を得る
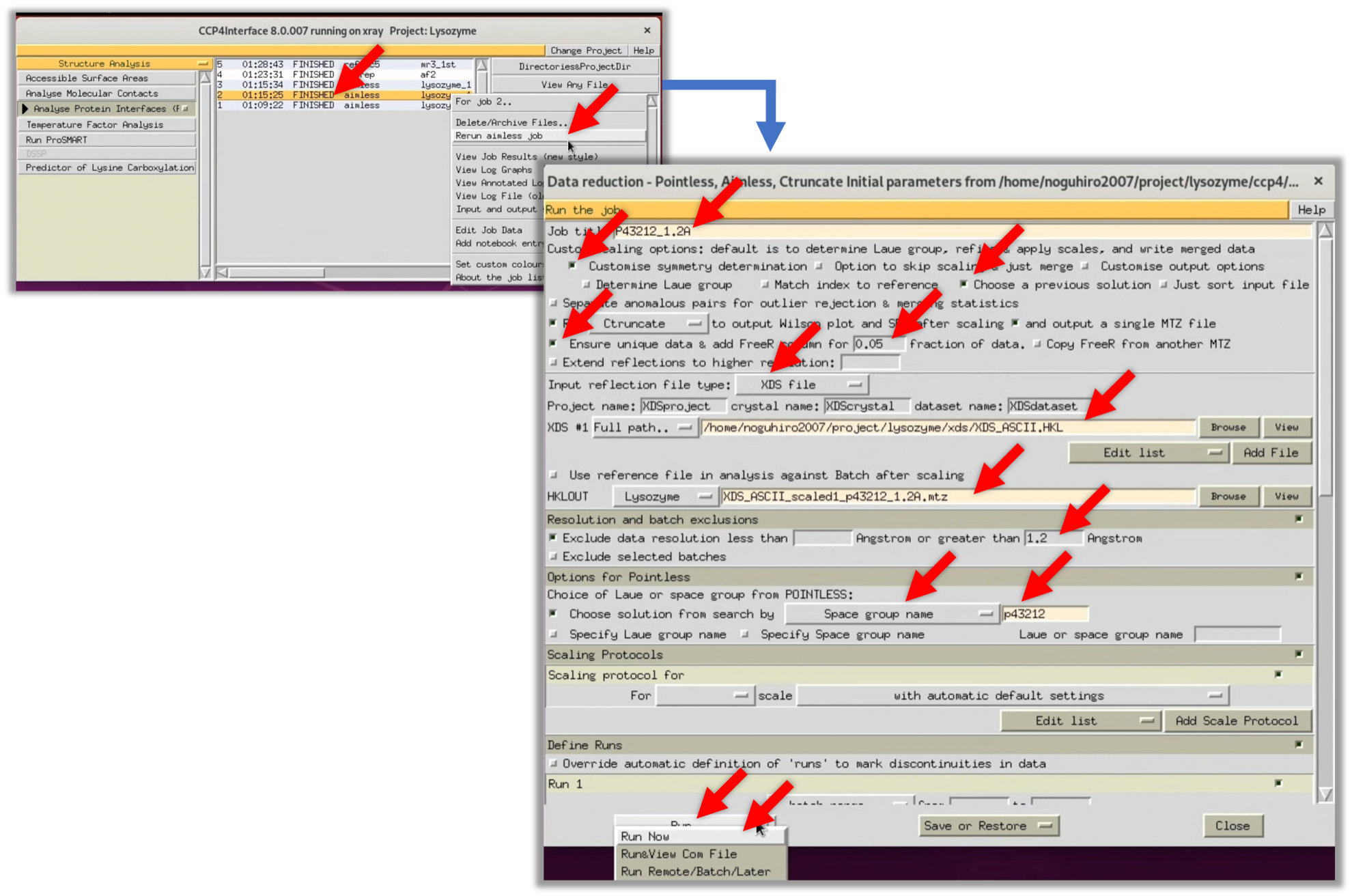
- 空間群P43212でScalingをやり直す(Step.2-5-2参照). パラメータの入力時間短縮のため, CCP4iのStatusのAIMLESS(P41212でScalingしたもの)を選択し, 右クリックで出てくる選択肢から
Return aimless jobを選び, 以前のパラメータ入力画面を開く. そして, 以下画像の赤矢印を参考に, まずは1.2Åの分解能・およびP43212のSpace group nameでScalingをやり直す.
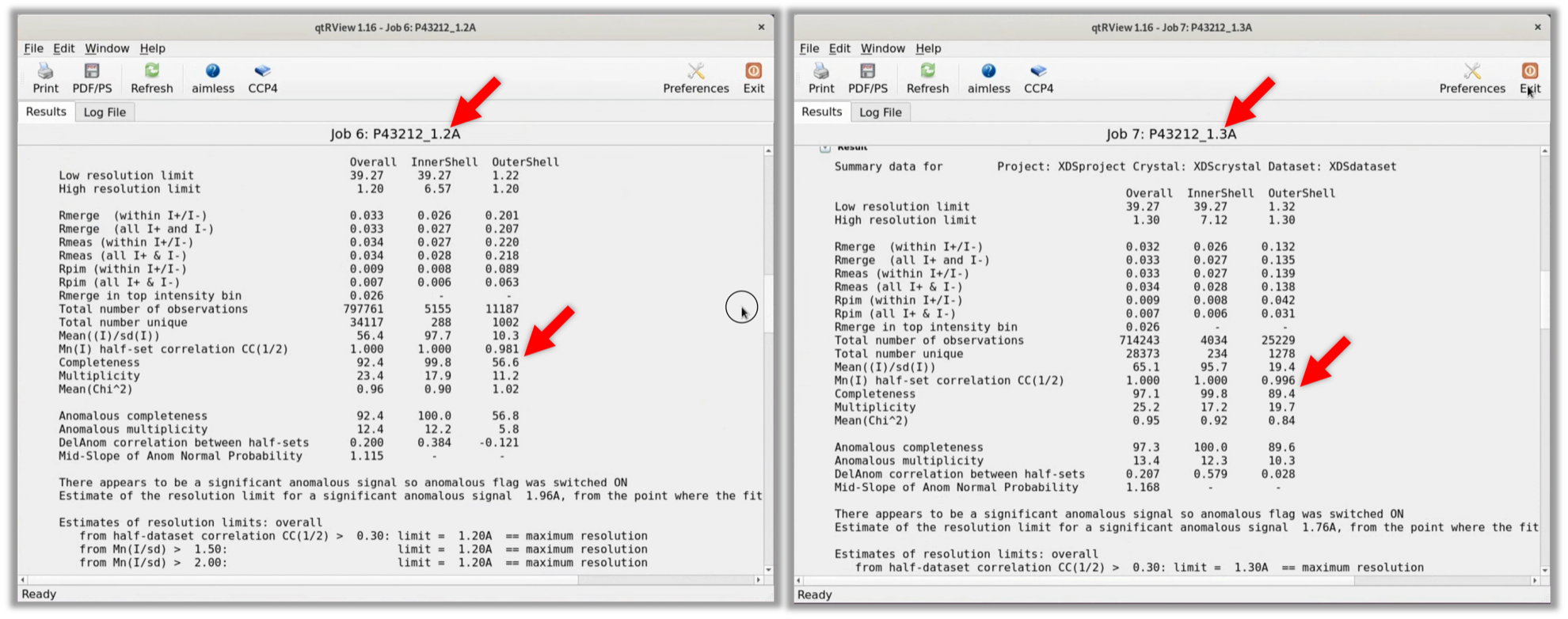
2. その結果, 空間群P43212/分解能1.2Åでは, OuterShellのCompletenessが56.6%であった. そのため, 空間群P43212/分解能1.3ÅでAIMLESSをやり直した結果, OuterShellのCompletenessが89.4%であったため, 空間群P41212で処理した際と同様に分解能は1.3Åと特定された.
Step.3-2-4 構造因子F(hkl)(空間群:P43212)を用いたMR法
前述, Step.3-2-1 構造因子F(hkl)(空間群:P41212)を用いたMR法と同様に空間群P43212の構造因子$F(hkl)$を用いて, MR法を行う.
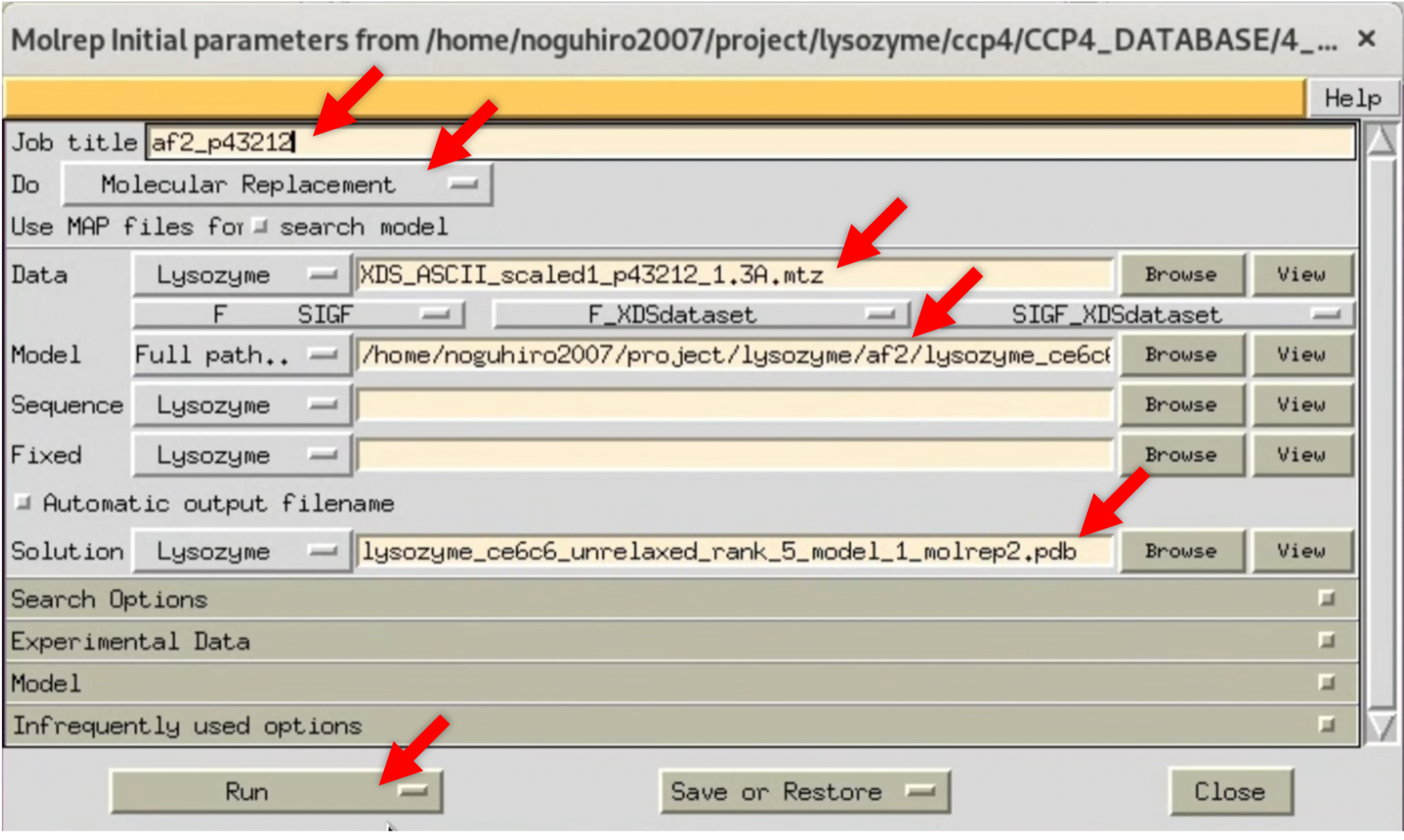
- Step.3-2-1 構造因子F(hkl)(空間群:P41212)を用いたMR法と同様にMolrepのパラメータ入力Windowを開き, 先にScalingした空間群P43212/分解能1.3Åの構造因子$F(hkl)$を用いてRunを行う.
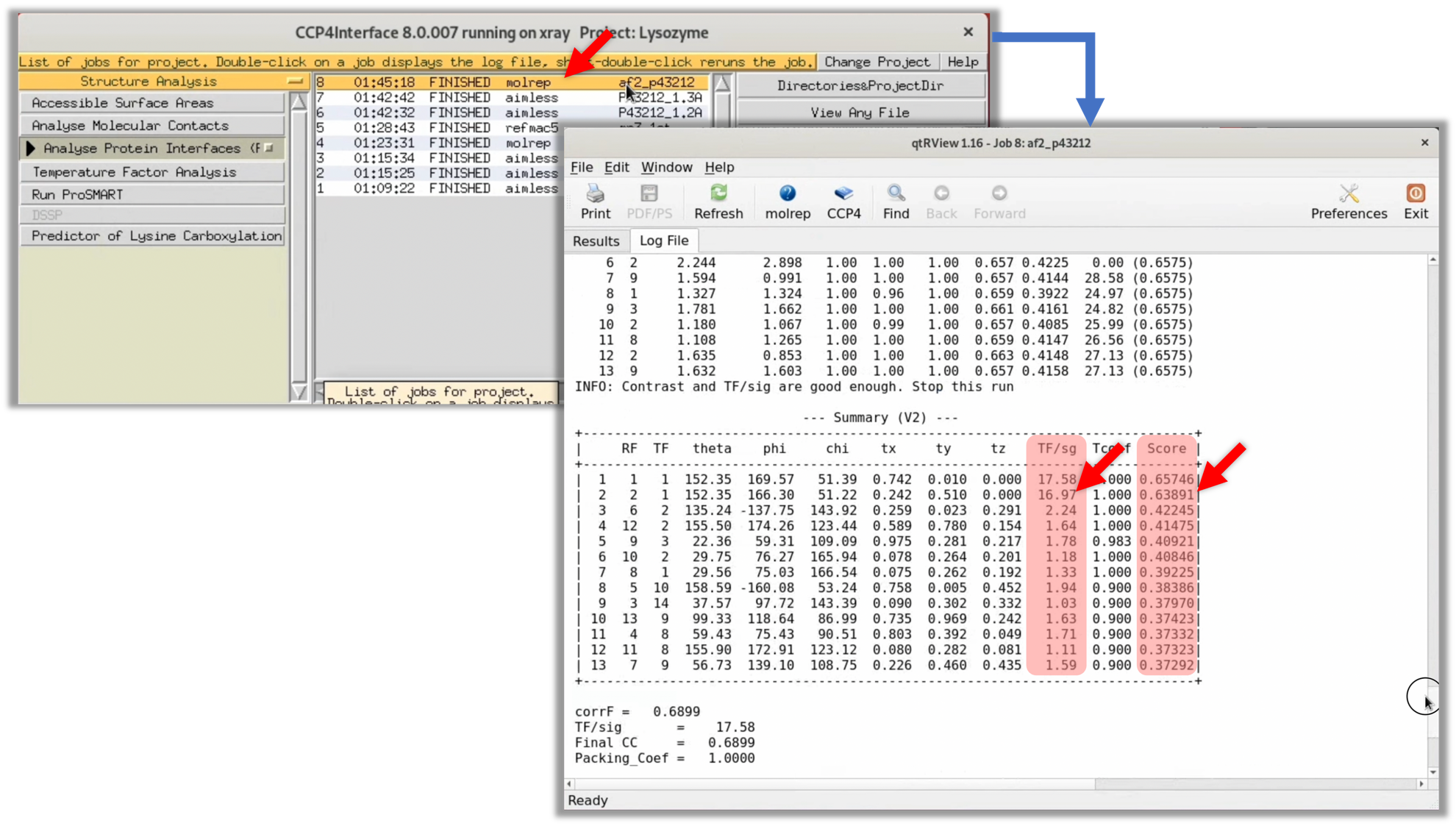
2. Molrepの処理が終了したら, そのLogを確認する. 結果, "Summary (V2)"の2行目のTF/sqおよびScoreが、3行目のそれと比較し優位に高く, Molrepは適切な初期位相を発見できた可能性が高い.
3. 確認のため, Step.3-2-1 構造因子F(hkl)(空間群:P41212)を用いたMR法と同様にRefmacを用いて電子雲マップ$\rho (xyz)$を作成し, Cootで見てみる. 以下の画像を参考に, Refmacのパラメータ設定を行いRunする.
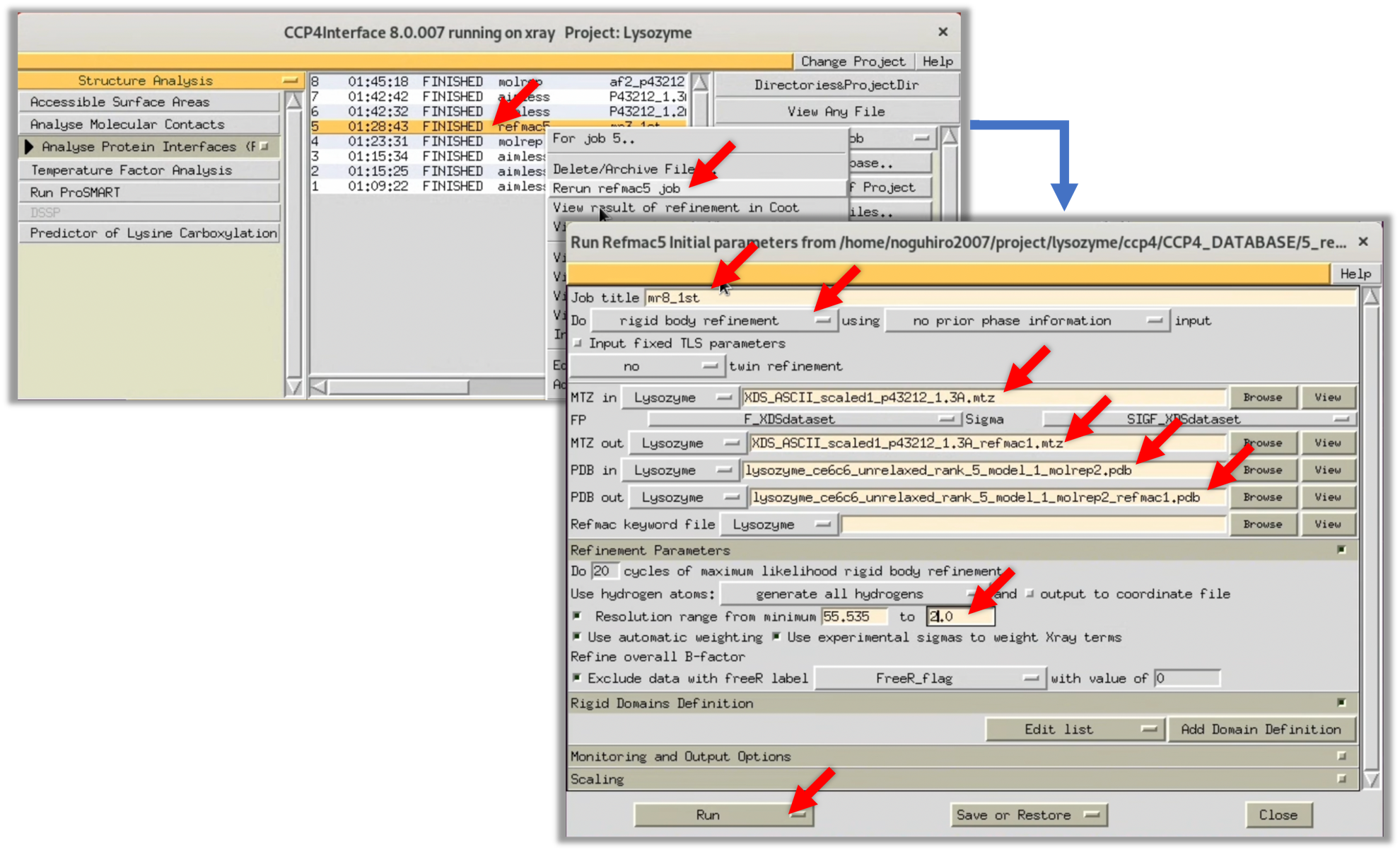
今回のRunでは, Refinement後のR-factor, R-free値(低い値ほどモデル構造と構造因子$F(hkl)$と合致していることを示す)は約40-41%であった. 完全なランダムを示す50%よりは低いが, 初期位相があっているかどうかを判断するには少し心もとない数字である.
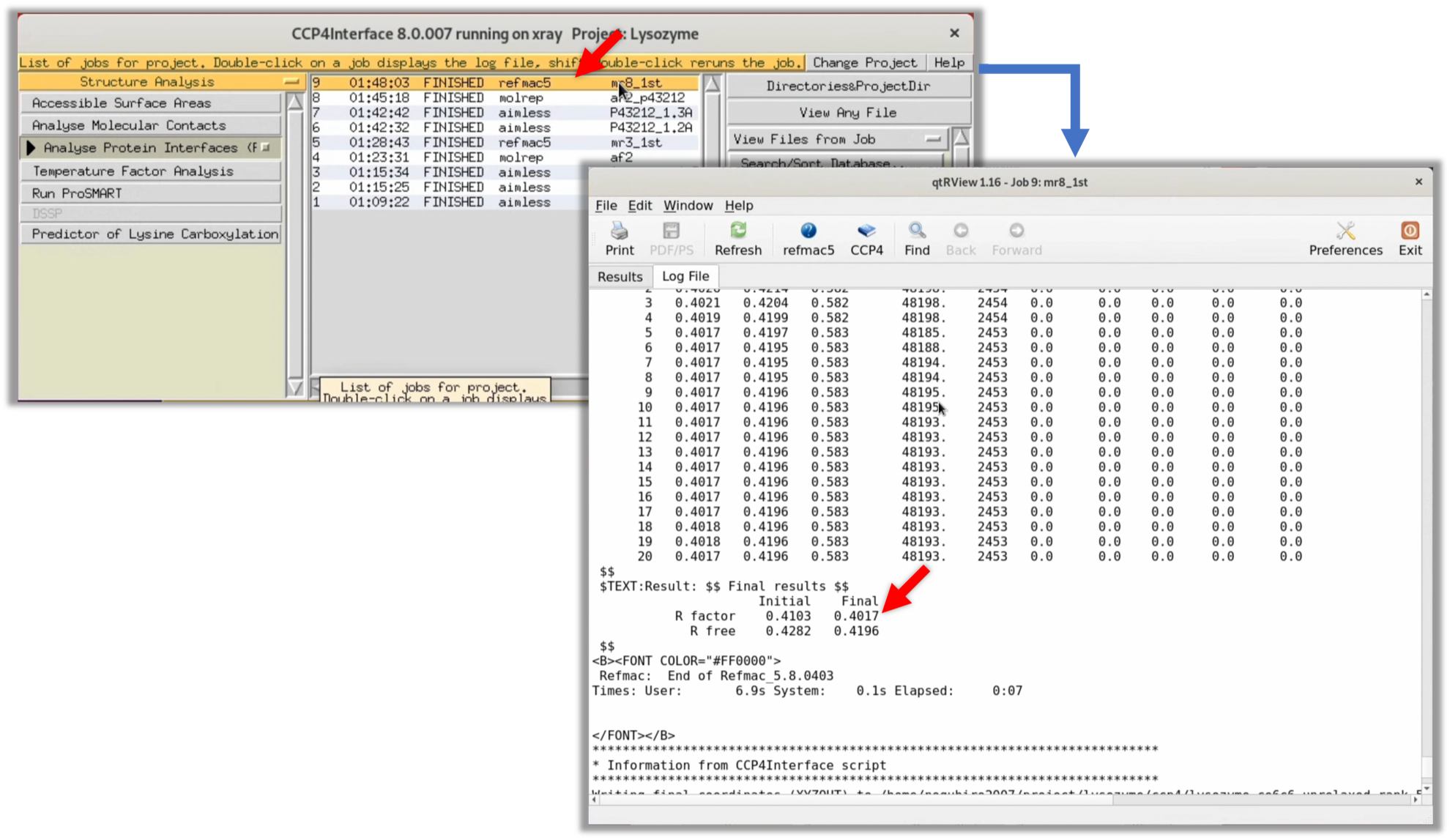
Step.3-2-1 構造因子F(hkl)(空間群:P41212)を用いたMR法と同様にRefmacのLogビューワーからCootを起動して電子雲マップ$\rho (xyz)$の確認を行った.
タンパク質モデルを全体表示した際, 隣接するLysozymeの間の溶媒空間と, Lysozymeそのものの電子雲マップ$\rho (xyz)$の濃度差をみる(動画:1:02 ~). 動画では電子雲マップ$\rho (xyz)$の可視化閾値を変化させても, 空間群P41212の際と比較し, より明確に両者の電子雲マップ$\rho (xyz)$が区別されている. よって, 良質な初期位相が得られていると考えられる.
電子雲マップ$\rho (xyz)$の目視確認でも判別がつかない場合, 構造精密化ステップまで進み, R-factor/R-freeの値を見て判断することもある.
著者が行った空間群P43212におけるMolrep, Refmacの処理結果をgithubにアップロードした. 適宜参考にしていただきたい.
Reference
- 分子置換法を用いた X 線結晶構造解析 (蛋白質科学会アーカイブ)
- タンパク質結晶構造解析入門 ―ブラックボックスの中身―: 分子置換法の原理と実際 (日本結晶学会誌, 2006 年 48 巻 5 号 p. 311-319)
- タンパク質結晶構造解析入門 ―ブラックボックスの中身―: 位相決定法の原理 (日本結晶学会誌, 2006 年 48 巻 4 号 p. 249-258)
あとがき
本タンパク質X線結晶構造解析チュートリアル第3回目では, 構造因子$F(hkl)$からAlphaFold2と分子置換法(MR法)を用いて初期位相を導出した. 次回, 最終回Step.4では構造精密化を行いLysozymeのモデル構造を構築してゆく.
他のStepへのリンク
- Step.0 構造決定の流れ
- Step.1 解析ソフトのインストール
- Step.2 回折画像の確認と構造因子の計算
- Step.3 初期位相を求める (本記事)
- Step.4 構造精密化とバリデーション