はじめに
AlphaFold2時代の構造解析チュートリアルシリーズの連載第4回目(初回, 第2回目, 第3回目).
今回は前回AlphaFold2の予測構造を用いて初期位相を出した構造を更に精密化し, 最終的なLysozymeのX線結晶構造を決定する.

目次
- Step.0 構造決定の流れ
- Step.1 解析ソフトのインストール
- Step.2 回折画像の確認と構造因子の計算
- Step.3 初期位相を求める
- Step.4 構造精密化とバリデーション (本記事)
この記事の対象と目的
タンパク質X線結晶構造法を専門外とする研究者・大学院生・学部生を対象とし, タンパク質X線結晶構造解析について回折画像から構造決定まで一通りできるようになることを目的とする. 前提として, Linux環境でBashコマンドをある程度打てる方を想定している.
本記事では, タンパク質の構造解析のチュートリアルを全4回の連載でお届けする. 今回は最終回, 第4回目である.
また, 本記事は完全にオープンである. 学校でも研究機関でも企業でも, ぜひこの記事を広めて, 教育に活用していただければ幸いである. しかし, 著作権は放棄していない.
タンパク質のX先結晶構造解析に必要な計算機環境
筆者の経験から, タンパク質のX線結晶構造解析には以下の計算機環境が適していると考えている. コンピューターパワーが必要であるため, 間違ってもRaspberry Piなどでやってはいけない.
- OS
- Ubuntu, CentOSなどのLinux (強く推奨)
- MacOSX
- Windows 10/11 (ネイティブ環境もしくはWSL2上のLinux環境を使用)
- CPU: > 4 core (推奨:> 16 core)
- Main Memory: > 16 GB (推奨: > 32 GB)
- Storage: > 50 GB (ソフトウェアインストールに約 12 GB.データセット 20 - 30 GB/crystal)
- GPU:必須ではない(推奨: GPU搭載)
本StepではStep.1で構築したGoogle Cloud Platform上の仮想マシン(Ubuntu 20.04LTE)で解析を行う.
Githubレポジトリ
本チュートリアルに関連する代表的なファイルはGithubにアップロードしている. 適宜ご参考いただければ幸いである.
Step.4 構造精密化とバリデーション
Step.4-0 準備
Step.4-0-1 構造精密化・バリデーションとは
Step.3で初期位相とともに得られたタンパク質Lysozymeのモデル構造は, AlphaFold2によって高精度に「予測」された構造を剛体として実験データに当てはめただけであり, アミノ酸単位で細かく見ると実験データと一致しないところがところどころ存在する. そこで, Step.4では構造精密化(Refinement)とバリデーションの作業を行い, タンパク質モデル構造を細かいところまで実験データと一致させ, タンパク質の最終的なX線結晶構造を得る. 構造精密化は下図の手順で行う. Step.3で初期位相が得られたモデル構造はまず, ①COOTで人の判断力を頼りにモデル構造を電子雲$\rho (xyz)$に合わせる. その後, ②構造精密化アプリケーションのRefmacを用いてモデル構造(各原子の座標と温度因子B-factor)から計算された構造因子$Fc(hkl)$(Fcのcはcalculatedのc)を, Step.2で得られた実験的な構造因子$Fo(hkl)$(Foのoはobtainedのo)に合うように, モデル構造の各原子位置(xyz)と温度因子B-factorを改良する. そして, 出力されたモデル構造を, FcとFoの一致度を示すR-factor/R-freeの値を参考に, さらにCOOTで構造の手直しを行う. このサイクルを複数回繰り返し, 最終的にタンパク質のモデル構造の品質チェック(バリデーション)で問題がないこととR-factor/R-free値が最小であることを確認し, 晴れてタンパク質のモデル構造が決定される.

ここで, R-factor値とR-free値について構造精密化に非常に重要であるため簡単に解説したい. R-factor(単にRと表記されることもある)およびR-freeは端的に言えば, モデル構造が実験的に得られた構造因子$Fo(hkl)$にどれだけ一致しているかを示した値(0に近いほど一致度が高い)であり, 以下で計算される.
\text{R-factor} = \frac{\sum ||Fo| - |Fc||}{\sum |Fo|}
しかし, シンプルで簡便なR-factor値のみを頼りに精密化を行うことには課題が存在する(RCSB: Learn: Guide to Understanding PDB Data: R-value and R-free
). 例えば, 本来タンパク質にあたる電子雲$\rho (xyz)$にすべて水分子のモデルを置けばR-factor値が低くなってしまい(=数値的に正しいと判断され), 本質的に間違った構造が得られてしまう. この問題を解決するため, クロスバリデーションの手法を用いたR-free指標を用いる. 具体的には, 構造因子$Fo(hkl)$から約5~10%のデータを検証用として除き, 残った約95-90%のデータを用いて精密化を行う. そして精密化後, 除かれた約5~10%の観測値を用いてR値を算出したものが, R-freeである. 仮に電子雲$\rho (xyz)$に100%フィットするタンパク質構造モデルを構築出来たのならば(現実では不可能であるが), R-free値はR-factor値とほぼ同じになる. 実際には, R-freeはR-factorよりも高い値をとる. 経験的にはR-factorより4%(0.04)程度高い値がちょうどよいと考える研究者もいる.
Step.4-0-2 構造精密化・バリデーションの戦略
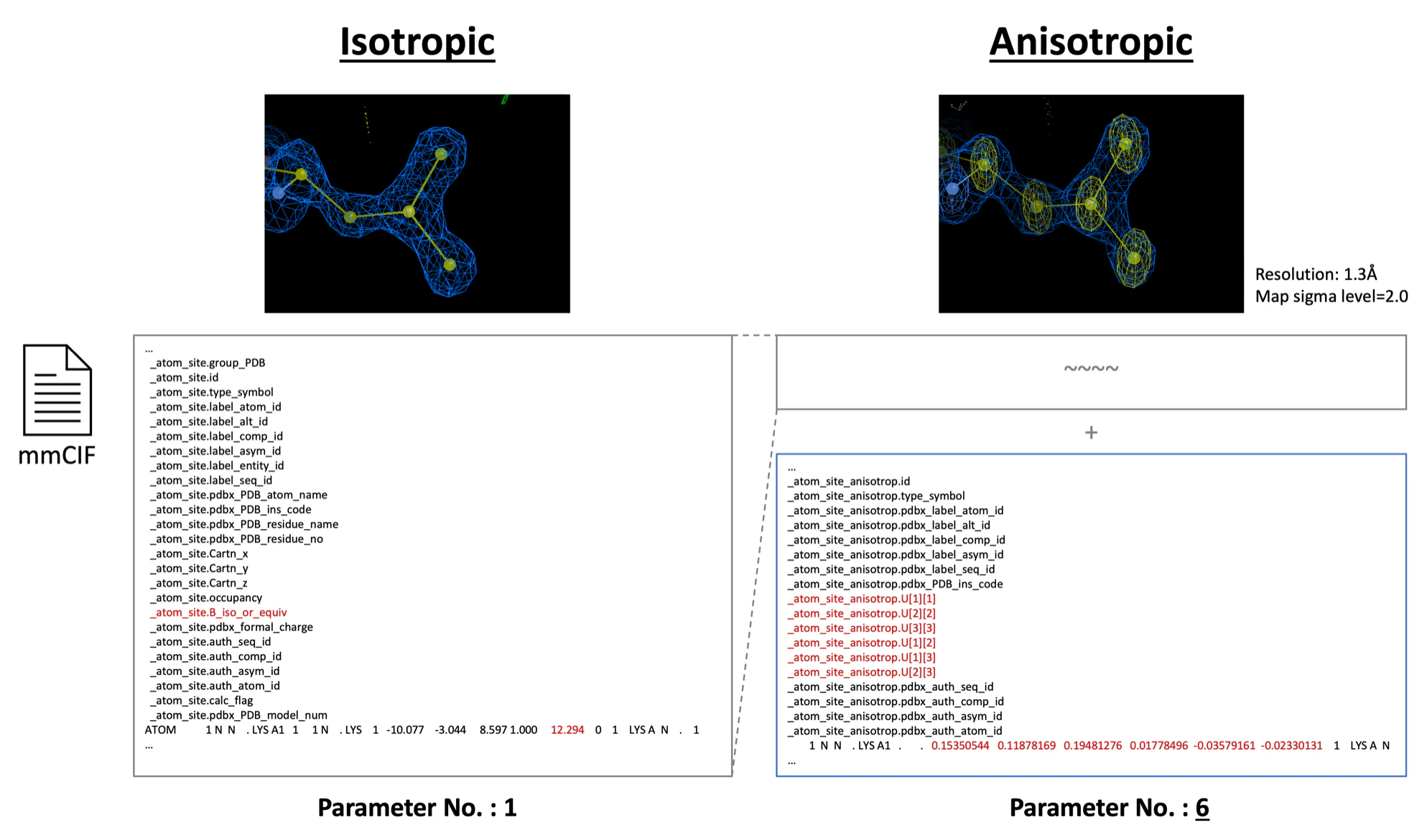
構造精密化は, 実験的に求められた構造因子$Fo(hkl)$に最も一致するモデル構造の要素(タンパク質分子・水分子・リガンド分子など)の原子座標$x,y,z$と温度因子B-factorを導出する工程である. この過程で初期位相$\alpha(hkl)$も最適化される (上記Step.4-0-1参考). 実際にPDBに登録されているLysozymeのmmCIFファイル1を見てみよう. mmCIFファイル内での原子の座標$x,y,z$と温度因子B-factorの示し方を, 下図に示す. 本チュートリアルで使用するLysozymeのタンパク質構造だけでも1,001原子あり, 各原子は4つのパラメータ(原子位置$x,y,z$と温度因子B-factor)を有するので, 総パラメーター数は4,004にも達する. 問題は, 実験で得られる構造因子$Fo(hkl)$のデータ量では, これらすべてのパラメーターに数理計算的に適切な解を与えることは不可能なことである2.

そのため, 実験で得られた構造因子$Fo(hkl)$に限りなくフィットする原子位置$x,y,z$と温度因子B-factorを求めるには戦略が必要になる. これは数値計算において適切な解を求める戦略と同様であり, 基本的には少ない入力データ量と少ないフィッティングパラメーター数からスタートして大まかな解を求め, 徐々に多くの入力データ量・多くのフィッティングパラメーター数に移行し詳細な解を得る..
この概念を以下の図に示した. いきなり多くの入力データ(今回の場合, 実験的に求められた構造因子$Fo(hkl)$の高分解能データ)で, 多くのパラメータを一気に適合させると最も適合する数値から外れた偽の解が局所解として得られてしまう危険性がある(最もフィッティング出来ている場合を青破線, 偽の数値が解として得られた場合を黒破線で示す)(A). Bではそれを回避するために, まずは少数の入力データと少数の変数を用いて大体の解を求め, 徐々に両者の量と数を増やしていくと, 局所解を回避して最適解をスムーズに求められる. 特に今回のような, 1.3Åを超える高分解能データの構造精密化では重要な戦略となる.

ここで一つの疑問が湧く. それは, 「たとえ図Aのように局所解が求められたとしても電子雲$\rho (xyz)$を見れば間違った解であることが分かるのではないか?そうならば, わざわざ手間をかけて図Bのような戦略を行わなくてもよいのではないか? 」ということである. しかし, それは 位相バイアス (Phase bias) と呼ばれる現象によって阻まれる. 専門誌Acta Crystallographica Section Dに掲載されたThe phase problemという論文に位相バイアスを端的に示す図が掲載されており, 非常に有用なので以下に引用する.
The Figure 3 of The phase problem. Garry Taylor, Acta Crystallographica Section D, Volume 59| Part 11| November 2003| Pages 1881-1890

この図ではアヒルの絵をフーリエ変換で擬似的に構造因子としたデーターに, 同様に猫の絵をフーリエ変換した際の位相情報を混ぜた絵を逆フーリエ変換して, 実空間で再び絵として示している. 本来であればアヒルの絵に猫の絵の位相情報が加えられているので, 絵の解釈は難しくなると予想されるが, 実際は猫の位相情報に大きく引っ張られて, 猫の絵が出てきてしまっている. これが位相バイアスと呼ばれる問題であり, 実際の構造精密化では間違って置かれた構造の位相情報に引っ張られ, 偽の電子雲が出現してしまう. 結果的に構造解析をして言える人間はこの状態に気づかず, 誤ったモデル構造を出力してしまう. 本問題は特殊な現象ではなく, 全体構造・局所的な構造に限らず構造精密化中に頻繁に見られ, 常に位相バイアスに留意して構造精密化を行う必要がある.
位相バイアスによって偽の電子雲$\rho (xyz)$が見られ, モデル構造と一致していたとしても, R-factor/R-free値は影響を受けず, 悪い値のままである. ここで研究者は間違いに気づけるが, この時点でモデル構造は偽の電子雲$\rho (xyz)$によくフィットしているため, 正しいモデル構造の位置がわからなくなってしまう. すなわち局所解に陥ってしまっており, リカバリーするには手間がかかる.
以上のことを踏まえ, 筆者が行ったLysozymeの精密化の概要を以下の図に示す. 今回は上記に示した構造精密化サイクルを11回繰り返すことで, モデル構造の決定を行うことが出来た. それぞれ以下に簡単なサイクルの解説を記す. ぜひ本チュートリアルをハンズオンされる際の参考にしてほしい.

| Cyc. No. | Comment |
|---|---|
| 1 | 分子置換法で得られたタンパク質構造全体(=AlphaFold2構造)を剛体として捉え(RBR, Rigid-Body Refinement), 分解能2.0Åのデータで構造精密化を行った. RBRは最もフィッティングパラメーターが少ない構成で行う精密化であり, 非常にラフな解を求めた. |
| 2 | 分解能は2.0Åそのままで, 精密化のパラメーター束縛法をRBRからRestrained Refinementに変更した(以下コラム参考). この変更で各原子が動ける自由度が上昇したことでR-factor/R-free値は大きく下がり, 本結晶の空間群が$P4_32_12$であることを改めて強く確信できた. |
| 3 | 引き続きRestrained Refinementと分解能で精密化を行った. Cyc.2と同様にR-factor/R-freeが下がったがその下がり幅は鈍化した. |
| 4 | 他の精密化パラメータは維持したが, 今回得られたデータの最高分解能(1.3Å)で精密化を行った.結果, R-freeは相当改善したが, R-factorはさほど変化しなかった. |
| 5 | ここで, 温度因子のパラメーターをIsotropicから, Anisotropicへ変更し, 大幅にフィッティングパラメーター数を増やした. これは得られた構造因子$Fo(hkl)$の分解能が1.3Åと高く, この分解能の電子雲$\rho (xyz)$をモデル構造で表現するにはAnisotropicが持つパラメータ数が必要なためである. しかしながら, あまりR-factor/R-free値は低下しなかったことから, タンパク質本体のモデル構造の構築はほぼ終了したと考えられた. |
| 6 | タンパク質モデル構造の周りに存在する水分子(構造水)を加えた. 結果, R-factor/R-free値が大きく低下した. |
| 7 | 6回目に引き続き, タンパク質構造と構造水を修正した. 結果, 前回に引き続き, R-factor/R-free値が大きく低下した. |
| 8 | このサイクルからCOOTのRamachandran PlotおよびGeometryバリデーションチェックツールを用いてタンパク質のモデル構造についてチェックを行った. |
| 9 | 8サイクル目と同様. 小規模な修正のみを行った. |
| 10 | 9サイクル目と同様. 小規模な修正のみを行った. R-factor/R-free値が落ち着いたため, タンパク質本体および構造水のモデル構築はほぼ完了したと考えられた. |
| 11 | 最後の仕上げとして, MolProbityを用いてLysozymeのモデル構造のチェック, およびモデル構造の修正・精密化を行い, 最終的にLysozymeのモデル構造を決定した. |
Refmac5の精密化メソッド
前述の通り, X線結晶構造解析のジレンマとして, 入力データ量に比べて最適化すべきパラメータ数(原子位置$x,y,z$と温度因子B-factor)が多すぎる問題がある. そのため, 各最適化パラメーターに既知の知見を活用した制限を与え, 適切なモデル構造の構築を行っている. その代表的な例として, Ridig Body Refinement(RBR), Restrained Refinement, TLS Refinementなどが考案されている. もちろん, 全く束縛条件を考えないUnrestrained Refinementも存在するが, かなりの高分解能(例: 1.0Å以上など)の条件でなければ信頼できる構造を得ることは出来ない. 以下に, 代表的な精密化メソッドについてそれぞれ簡単に解説する.
- Rigid Body Refinement (RBR): 剛体精密化とも呼ばれる. モデル構造のサブユニット(chainごと)を一つの剛体として捉え, サブユニット全体の位置と向きを表す6つの自由度(3つの平行移動と3つの回転)を最適化する. 主に, MR法で初期位相を求めたあとの構造精密化の初期段階で使用され, 非常に粗い解を求めるために用いられる.
- Simulated Annealing Refinement (SA): 構造モデルを擬似的に高温に加熱して徐々に冷却することで, モデル構造が局所解に陥るのを避ける方法である. 今回のTutorialでは実施しなかったが, Ribid Body Refinementの後に行っておくと良い.
- Restrained Refinement: これまでに報告されてきたタンパク質の超高分解能構造や化学的な制約が明らかにしたタンパク質構造が本質的に持つ構造的特性を考慮して, 各原子間に束縛条件(立体化学的束縛:共有結合, 角度, 二面体, 平面性, キラル性, などを反映)を加えることで, 実質的にパラメータ数を減らして精密化を行う手法である. 通常のタンパク質構造精密化で得られる1.3 - 3.0Å程度の分解能では, Restrained Refinementで構造決定することが多い.
- TLS refinement: 各原子の温度因子B-factorを, モデル構造のサブユニット(chainごと)の回転, 並進, 振動と組み合わせて表現して精密化を行う方法である. 各原子の温度因子B-factorの相関を考慮することで, 構造モデルの精度と信頼性を高めることができる. 分解能が低い場合や, 特に原子の温度因子B-factorが高い場合(=結晶が結晶中で動きが大きい)場合に有効である. 通常用いる精密化メソッドではなく, 必要がある際に用いる.
- Unrestrained Refinement: 文字通り, 原子座標($x,y,z$)や温度因子B-factorを, 一切の束縛条件なしに観測された構造因子$Fo(hkl)$との一致度を最大化するように行う精密化法である. 一番シンプルだが, 超高分解能(例: 1.0Å以上)が必要で, タンパク質X線結晶構造解析における適用事例は多くない.
IsotropicとAnisotropic
各原子の温度因子($B-factor$)の表現方法の違いを指す. Isotropicは原子の温度因子(B-factor)を方向に存在しない1つの値で示す. もちろん科学的事実として各原子の電子軌道は円ではないが, 多くのタンパク質X線結晶構造解析で得られる分解能(1.3~3.0Å程度)では, 各原子の電子雲の正確な形を観察することは出来ず, 球状であると仮定している. 一方, 1.3Å以上の高分解能(基準は研究者によって見解は異なる)では, データ量が多く, 各原子の温度因子(B-factor)の非対称性や相関性をより正確に表現できるため, 方向に依存する6つのパラメーター(U-factors)で示すことが可能である. 以下の図では実際のmmCIFファイル上でどのように表現されているかを示した. Isotropicでは一つの温度因子パラメータ(B-factor)しか存在しないが, Anisotropicでは6つの温度因子パラメータ(U-factors)が記録されている.
電子雲$\rho (xyz)$の種類
これまで電子雲$\rho (xyz)$と単純に示してきたが, 実はタンパク質のX線結晶構造解析で用いられる電子雲には主に2種類あり, 我々が通常"電子雲$\rho (xyz)$"と言ったときに指す2Fo-Fcマップと, Fo-Fcマップである. COOT上では, 下図Aのように2Fo-Fcマップは青色のマップで示され, Fo-Fcマップは緑/赤色で示される(緑はpositive densityを示し, 赤がnegative densityを示す). 構造精密化ではこの2つのマップを要所要所で適切に用いて構造精密化を行う.
下図Aの場合, アミノ酸側鎖の配向がFo-Fcがマイナスのマップに存在しており, この配向は実験データと合っていないことが分かる. よって, 下図BのようにFo-Fcのポシティブマップ似合うように訂正する.

以下, 簡単に2つのマップを説明する.
- 2Fo-Fcマップ : 実験から得られた構造因子$Fo(hkl)$を2倍し, そこからモデル構造から計算された構造因子$Fo(hkl)$を引いてマップを計算したのが, 2Fo-Fcマップである. 計算式から明らかなように, モデルがきちんと置かれていれば通常濃度の電子雲マップが出現するが, 仮にそこにモデルが置かれていなければ強調された電子雲マップが出現する.
- Fo-Fcマップ : 実験から得られた構造因子$Fo(hkl)$から, 単純にモデル構造から計算された構造因子$Fo(hkl)$を引いたのが, Fo-Fcマップである. Fo > Fcの場合はpositive density map(Fo-Fcが正の値)と呼ばれ, ”そこに何かがある"ことを示す(COOT初期設定では緑色で表示される). Fo < Fcの場合はnegative density map(Fo-Fcが負の値)と呼ばれ, "そこにモデル構造を置くのは間違っている"ことを示す(COOTの初期設定では赤色で表示される).
Step.4-1 構造精密化
では実際に構造精密化を行う. その準備としてモデル構造の修正に用いるCOOTの簡単な使い方と, 非対称単位(ASU, Asymmetric Unit)について解説する.
Step.4-1-0 COOTの操作方法と非対称単位(ASU)
Step.4-1-0-1 COOTの操作方法
以下にCOOTを使うための操作方法を示す. 基礎的な操作となるため, 精密化を行う前にぜひ一通り試していただきたい. なお, マウスの第3ボタンもフルに使うため, タッチパッドではなくマウスを推奨したい.
| 動作 | 操作 | COOTの動き |
|---|---|---|
| 回転 |
 左クリック |
 |
| 平行移動 (xy軸) |
 or  Ctrl+左ドラック/第3ボタンドラック |
 |
| ズーム |
 右ドラック (上下) |
 |
| 平行移動 (z軸) |
 Ctrl+右ドラック (上下) |
 |
| 次のアミノ酸/水/リガンドへ移動 |
 Space |
 |
| 特定原子へ移動 |
第3ボタン (原子選択) |
 |
| 電子雲マップ (奥行き) |
Ctrl+右ドラック (左右) |
 |
| 電子雲マップの等高線レベル変更 |
スクロールホイール |
 |
Step.4-1-0-2 非対称単位(ASU)とは
Cootを使ってタンパク質分子を視覚化するとき, 表示されるのは非対称単位(ASU, Asymmetric unit)です. タンパク質構造だけに限らず, 「結晶」は分子が特定のパターンで並び重なり形成されていますが, この結晶を最小限で表現する単位が非対称単位(ASU)となります. これを簡単に説明すると, 非対称単位(ASU)により結晶の構造単位, つまり単位胞(Unit Cell)が形成され, これらの単位胞が規則正しく空間を埋めることで結晶が形成されます.
特筆すべきなのは「非対称単位(ASU) ≠ 生物学的集合体(Biological Unit)」という事実です. つまり, Cootで表示したタンパク質構造がそのまま実際の環境で機能しているわけではないということです. 例えば, 約1.2万年前に絶滅したマンモスの巨体に酸素を運んでいたヘモグロビンタンパク質(Woolly Mammoth Hemoglobin)は, αタンパク質とβタンパク質がそれぞれ2つずつ, 合計4つで機能を発揮(=生物学的機能集合(Biological Unit))します. しかし, PDBに登録されている構造(専門的にはR構造, 3VRF)では, αタンパク質とβタンパク質がそれぞれ1つずつで構成される非対称単位(ASU)となっています.
なお, 非対称単位(ASU)の大きさは結晶そのものにより異なります. 実際, 先に述べたWoolly Mammoth Hemoglobinの異なる構造(専門的にはT構造, 3VRE)では, 非対称単位(ASU)と生物学的集合体が一致しています.
詳しくは各リンク先で, PDBに登録された構造と生物学的集合体(Biological Unit)を比較してみてください.
Step.4-1-1 Cycle.1
| Parameter | Value |
|---|---|
| Refinement method | RBR |
| Resolution | 2.0Å |
| B-factor | Isotropic |
| Target | Protein Structure |
実は精密化の1st cycleは前回のStep.3-2-4 構造因子F(hkl)(空間群:P43212)を用いたMR法の3ですでに行っている. 一番最初の精密化サイクルは, このようにMR法の確認のために行われる場合が多い.
動画で確認するように, 精密化(及びMR法)はうまくいっている.
Step.4-1-2 Cycle.2
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 2.0Å |
| B-factor | Isotropic |
| Target | Protein Structure |
RBRで精密化した後は, タンパク質構造のすべてのアミノ酸残基をチェックしてゆき, 主に主鎖/側鎖が電子雲$\rho (xyz)$から外れていないかをチェックする. 本チュートリアルでは, Lysozymeという定番の構造をAlphaFold2で高精度に予測し, 非常に良質な構造因子$F(hkl)$に当てはめていることから, RBRの時点で大まかに一致していると判断(じっさいにしている)し, COOTを用いた構造修正の前に一回Restrained Refinementを行った(勘所がある方以外は次のCycle.3の内容をここで行われたい).
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-3 Cycle.3
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 2.0Å |
| B-factor | Isotropic |
| Target | Protein Structure |
Cycle.2で電子雲$\rho(xyz)$を改善したので, 人の手で構造を修正してゆく. その様子を以下の動画(5倍速)で示す. このように, タンパク質chainのN末端からC末端まで, 1残基づつ丁寧に確認・改善してゆく. 途中, 主鎖および側鎖が電子雲マップ(2Fo-Fc及びFo-Fc両マップ)と合っていない部分があれば, その部分をReal Space Fittingで合わせてゆく. 長い領域で電子雲マップとモデル構造が合っていなければ, 一度モデル構造を削除(COOT右側のゴミ箱アイコンが削除機能である)してから, 新たにタンパク質主鎖構造を構築(COOT右側の+マークにアミノ酸1残基分が記されたアイコンが追加機能である)したほうが効果的にモデル構築を行える場合もある.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).
Step.4-1-4 Cycle.4
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 1.3Å |
| B-factor | Isotropic |
| Target | Protein Structure |
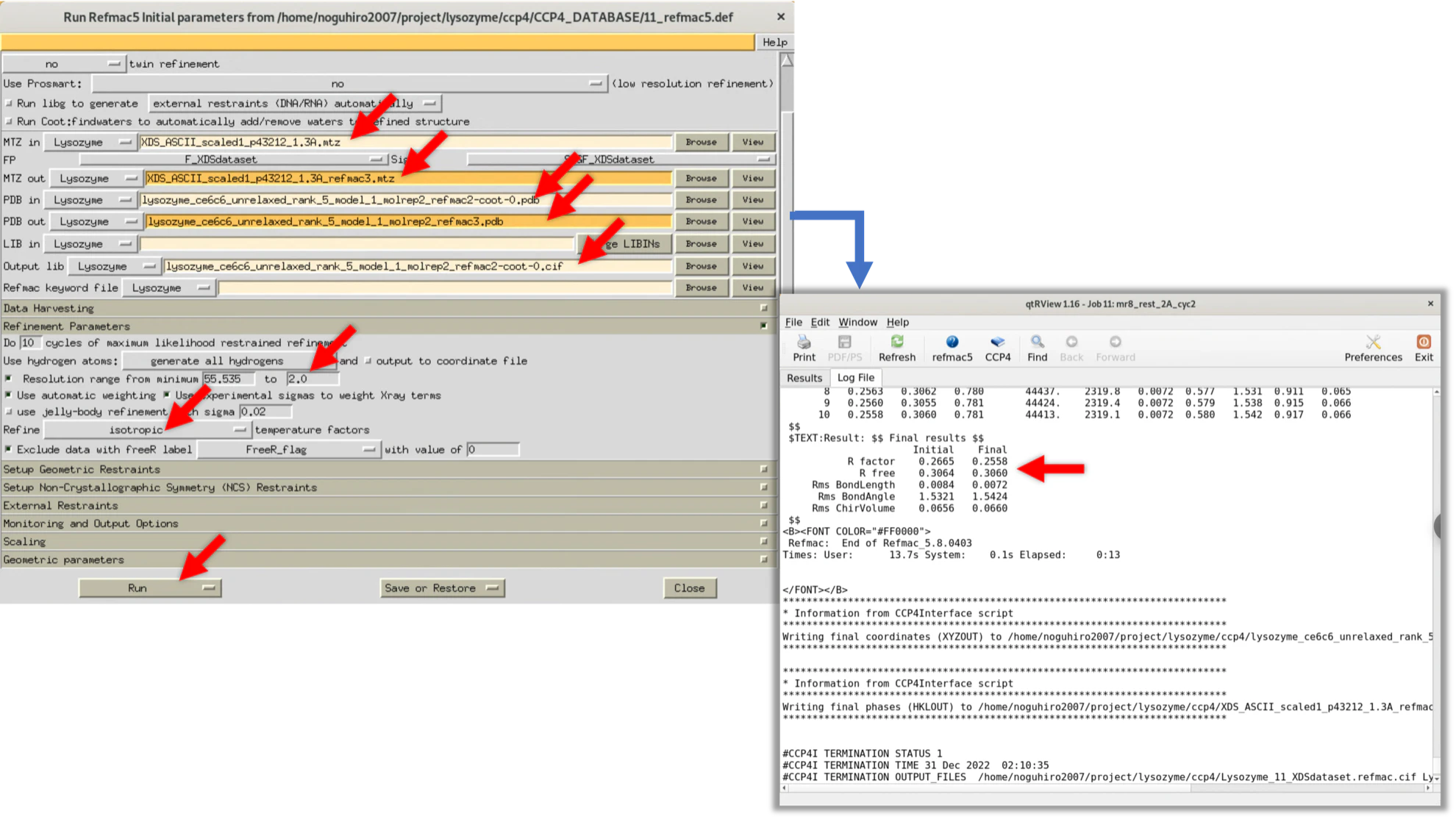
Cycle.3で修正した箇所を確認し, 再び電子雲マップと合っていない箇所については修正を行う. 筆者が行った際は, この時点で電子雲とLysozymeのモデル構造の一致度が高かったため, Refmac実行時には分解能1.3Åの構造因子$Fo(hkl)$を用いた. 結果, R-factor/R-free=0.258/0.271となり, R-freeが大きく低下した.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-5 Cycle.5
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 1.3Å |
| B-factor | Anisotropic |
| Target | Protein Structure |
これまでの分解能2.0Åのときと同様に, COOT上で分解能1.3Åの電子雲マップにタンパク質モデル構造をあわせた. この段階になると, ほぼ側鎖も含めて一致している. そのため, Isotoropicを用いた精密化は一段落したと判断し, 分解能1.3ÅだからこそできるAnisotropic精密化を行った. 結果, R-factor/R-freeともにある程度低下し, Anisotropicによるパラメーター増加によってよりフィッティングが改善されたことが示された. しかし, 下げ幅が小さかったためタンパク質本体の精密化はほぼ完了したと考えられた.
実際のCOOTを用いた構造修正については, 以下の動画(x5倍速)を参考にしていただきたい.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-6 Cycle.6
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 1.3Å |
| B-factor | Anisotropic |
| Target | Protein Structure + Water |
タンパク質のモデル構造の周りに存在する水分子(構造水)を加えた. 水分子(構造水)はCOOTでBlob(電子雲マップの塊)全てに一つ一つ手動で割り当ててもよいが, 数百個ほどある水分子全てにそれを行うのは非現実的である. 故に自動で割り当てる方法がいくつかあるが, 今回はその中からCOOTによる自動割当を行った. やり方は動画を参照されたい.
Refmac処理の結果, R-factor/R-free値が大きく低下した. このように高分解能になるほどタンパク質の周りに存在する水分子(構造水)がはっきりと確認でき, 構造全体への寄与も無視できなくなる.
構造水のピックアップ基準と戦略
構造水とは結晶中でタンパク質と水素結合している水分子を指し, 位置がある程度固定化していることから電子雲マップで視認可能である. タンパク質の安定性解析や活性部位解析等を行う際に非常に重要である. タンパク質に直接水素結合しているものを一次構造水, 一次構造水と水素結合して直接タンパク質分子と分子と結合しない構造水を二次構造水と呼ぶ.
タンパク質モデル構造本体の精密化と同様に, 構造水の精密化も大局的-->局所的という流れで行い, 全体として局所解に陥るのを防ぐ. また, ノイズを拾ってしまうことを防ぐため, 一定以上の電子雲マップ濃度で確認されたBlobのみ, 構造水としてピックアップする.
以下に著者が構造水をピックアップするための基準を示す.
| Parameter | Value |
|---|---|
| H-bond distance | 2.2 - 3.5 Å |
| Pickup Sigma Level | > 1.1 $\sigma$ (2Fo-Fc) |
まずは, Pickup Sigma Levelの高いSigma(例えば2.0$\sigma$)からはじめ, マップが強く出ている構造水からピックアップしてゆき, 徐々に1.1$\sigma$にPickup Sigma Levelを下げてゆく手法を筆者はよく用いる. 是非参考にしていただきたい.
実際のCOOTを用いた構造修正については, 以下の動画(x5倍速)を参考にしていただきたい.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-7 Cycle.7
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 1.3Å |
| B-factor | Anisotropic |
| Target | Protein Structure + Water |
Cycle.6に引き続き, タンパク質構造と構造水を修正した. 結果, 前回に引き続きR-factor/R-free値が大きく低下した. これは, 前回の構造水を加えたことで周辺の位相が変化し, これまで見えなかった新たな構造水の電子雲が見えるようになったためである. また, 二次構造水も多数確認されている.
実際のCOOTを用いた構造修正については, 以下の動画(x5倍速)を参考にしていただきたい.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-8 Cycle.8
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 1.3Å |
| B-factor | Anisotropic |
| Target | Protein Structure + Water + Ramachandran Plot + Geometory check |
このサイクルからタンパク質モデル, 構造水に加え, COOTのRamachandran PlotおよびGeometryバリデーションチェックツールを用いてタンパク質モデル構造についてチェックを行った. いずれも, 過去に高分解/能超高分解能で構造解析されたペプチドや小さいタンパク質分子といった信頼できる実験データを元にタンパク質がとり得る構造的特徴を定め, タンパク質モデルの構造的信頼性を評価するものである.
Ramachandran Plotとは
1963年にG. N. Ramachandran, C. Ramakrishnan, V. Sasisekharanによって開発されたもので, タンパク質構造中のアミノ酸残基のφとψの二面角に対するエネルギー的に許容される領域を視覚化する方法である(Wikipedia).

理論的に, アミノ酸残基がタンパク質中で取り得るφとψの角度(またはコンフォメーション)を示すために使用されるほか, これまでに測定された高分解能のX線結晶構造解析などのデータから, タンパク質の構造が許容されると考えられる範囲を経験的に検証するためにも用いられる. そのため, 時代とともにFavored/Allowed/Outilersの境界線はアップデートされており, 最新のCOOTを用いれば最新の境界線情報のもとでモデル訂正が可能である.
古典的なRamachandran Plot. Wikipedia: ラマチャンドランプロットより引用

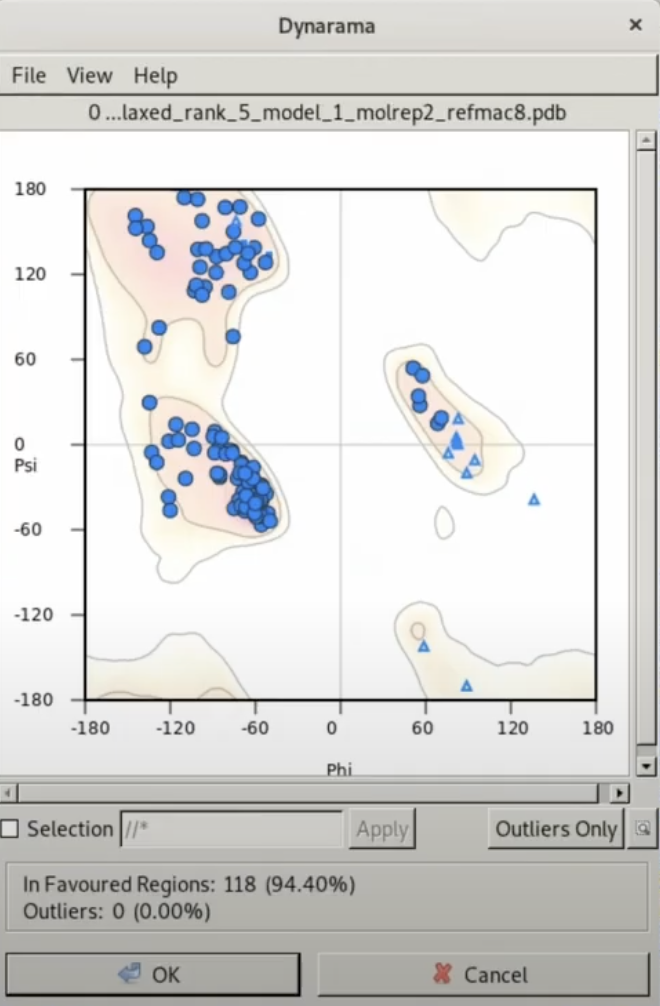
本チュートリアルで使用するCOOTに内蔵されているRamachandran Plotの例. 古典的なRamachandran plotと比較してFavored/Allowed/Outilers境界線の解像度が上がっている.
なお, Ramachandran PlotのFavored内にモデル構造を必ず収めなければいけない, というわけではない. これらはあくまで全タンパク質の統計的平均を示したものに過ぎず, 今回のように高分解能で電子雲がくっきり視認できるなど, 実験的根拠があればOutilersにあっても信頼できる構造と言える.
実際のCOOTを用いた構造修正については, 以下の動画(x5倍速)を参考にしていただきたい.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-9 Cycle.9
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 1.3Å |
| B-factor | Anisotropic |
| Target | Protein Structure + Water + Ramachandran Plot + Geometory check |
Cycle.8と同様に, タンパク質モデル構造本体と結合水, そしてタンパク質モデル構造のRamachandran PlotやGeometory checkを参考に構造の修正を行った. 一度構造を修正してRefmacで精密化をしたとしても, Refmac処理中にモデル構造が変化し, Geometoryが修正前に戻ってしまう場合もあるので注意が必要である.
実際のCOOTを用いた構造修正については, 以下の動画(x5倍速)を参考にしていただきたい.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-10 Cycle.10
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 1.3Å |
| B-factor | Anisotropic |
| Target | Protein Structure + Water + Ramachandran Plot + Geometory check |
Cycle.9と同様に, 小規模な修正を行った. また, 修正を繰り返す中で, R-factor/R-free値が落ち着いてきたため, 求めたい最適なフィッティングがなされていると判断した.
実際のCOOTを用いた構造修正については, 以下の動画(x5倍速)を参考にしていただきたい.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-11 Cycle.11
| Parameter | Value |
|---|---|
| Refinement method | Restrained Refinement |
| Resolution | 1.3Å |
| B-factor | Anisotropic |
| Target | Protein Structure + Water + Ramachandran Plot + Geometory check |
| Validation | Mol Probity |
最終的に, MolProbityを用いてLysozymeのモデル構造の総合的なチェックを行い, それを元にモデル構造を修正・精密化を行うことで最終的なタンパク質モデル構造を決定した.
MolProbityは代表的なタンパク質モデル構造のバリデーションツールである. CCP4と並ぶ構造解析ソフトウェアスイートであるPhenixの精密化ソフトであるPhenix.refineでは, 精密化後に自動的に内蔵されたMolProbityによる構造チェックが行われる. 一方で, 今回チュートリアルに用いてるCCP4のRefmac5の場合, 残念ながらそのような機能は搭載されていないため, 以下の手順でWebバージョンMolProbityによる構造チェックを行う. タンパク質モデル構造をPDBへ登録する際は必須の作業となる.
-
Web版のMolProbityにアクセスし, Cycle.10で出力されたPDB (or mmCIF)ファイルをアップロードする. そして, 画像右下の画面に到達する.

-
"Analyzer geometry without all-atom contacts"をクリックすると分析の前処理が開始され, 画像左下の解析項目の選択画面に遷移する. ひとまず, すでに選択されている標準的な解析項目で解析を行う. "Run programs to perform these analysis"をクリックし, 解析を行う.

-
解析結果が出てくる. Summary statisticsでは緑色ハイライトの項目が問題が無いことを示している. 一方, 赤色ハイライトの項目は, 問題があることを示しているため, 下にスクロールし, 何が問題かを把握する.

このように得られたMolProbityの結果を見つつ, COOTで構造を確認・修正してゆく.
実際の作業については, 以下の動画(x5倍速)を参考にしていただきたい.
Refmacの設定値と, 実行時のLogを載せる(筆者実施時の値. 同程度の値であれば問題ない).

Step.4-1-12 タンパク質のモデル構造決定
回折画像確認からここまで長い道のりだったが, MolProbityのバリデーション情報を元に構造を修正し, Refmacで最後の精密化を行うことで, ついに最終的なタンパク質のモデル構造を決定することができた.
決定された構造をPDBに登録する際や論文に掲載する際, 業界の中で通称”Table.1”(テーブルワン)と呼ばれる構造の統計値をまとめた表を作成する決まりがある. チュートリアルである今回はPDBや論文にするわけではないが, せっかくなので最後の仕上げまで行ってみる. Table.1は作成者によって掲載する項目が異なる場合が多い. よって, 以下に示す数値はあくまで筆者の流儀, ということをご了承いただきたい. 作成項目に迷った際は, 専門誌であるIUCr Acta Crystallographica Section Dに掲載されている論文のTable.1を真似ると良いだろう.
以下, 本チュートリアルで解析したLysozymeのTable.1を筆者のデータを元に記す. ぜひ参考にしてほしい.
| Lysozyme | 解説 | |
|---|---|---|
| Data collection | ||
| Diffraction source | NSLS-II 17-ID-2 | Step.2:使用した回折点データセットの情報より引用 |
| Wavelength(Å) | 0.97894 | Step.2:使用した回折点データセットの情報より引用 |
| Resolution range(Å) | 39.27-1.30 (1.32-1.30) | Step.2:Aimlessの結果(log)より引用 |
| Space group | P43212 | Step.2:Aimlessの結果(log)より引用 |
| a,b,c(Å) | 78.54, 78.54, 37.28 | Step.2:Aimlessの結果(log)より引用 |
| α, β, γ (°) | 90, 90, 90 | Step.2:Aimlessの結果(log)より引用 |
| Reflections (measured/unique) | 714,243/28,373 | Step.2:Aimlessの結果(log)より引用 |
| Completeness (%) | 97.1 (89.4) | Step.2:Aimlessの結果(log)より引用 |
| Mean $I / \sigma(I)$ | 65.1 (19.4) | Step.2:Aimlessの結果(log)より引用 |
| Multiplicity | 25.2 (19.7) | Step.2:Aimlessの結果(log)より引用 |
| $R_{pim}$ | 0.009 (0.042) | Step.2:Aimlessの結果(log)より引用 |
| CC(1/2) | 1.000 (0.996) | Step.2:Aimlessの結果(log)より引用 |
| Wilson B factor (Å$^2$) | 10.762 | Step.2:Aimlessの結果(log)より引用 |
| Refinement statistics | ||
| Resolution range(Å) | 35.15-1.30 | Step.4:最終モデル構造(mmcif,pdb)より引用 |
| R-factor (%)/$R_{free}$(%) | 13.16/15.65 | Step.4:最終モデル構造(mmcif,pdb)より引用 |
| No. of atoms in structure | ||
| Protein | 1,187 | Step.4:最終モデル構造(mmcif,pdb)より引用 |
| Ligand | 0 | |
| Water | 159 | Step.4:最終モデル構造(mmcif,pdb)ファイルのHOH行番号を参考に計算した値を引用 |
| R.m.s deviations from ideal | ||
| Bond lengths (Å) | 0.011 | Step.4:最終モデル構造(mmcif,pdb)より引用 |
| Bond angles(°) | 1.78 | Step.4:最終モデル構造(mmcif,pdb)より引用 |
| Chiral volumes(Å$^3$) | 0.114 | Step.4:最終モデル構造(mmcif,pdb)より引用 |
| Ramachandran plot, residues in (%) | ||
| Most favorable region | 98.5 | Step.4:最終モデル構造のMolProbityの分析結果から引用 |
| Additional allowed region | 100.0 | Step.4:最終モデル構造のMolProbityの分析結果から引用 |
| Average B factor(Å$^2$) | 13.559 | Step.4:最終モデル構造(mmcif,pdb)より引用 |
(Values in parentheses are for the outer shell.)
Table.1の作成をもって, 構造精密化は終了となる. 大変お疲れさまである.
著者が行ったRefmac5による構造精密化の結果をgithubにアップロードした. 是非参考にしていただきたい.
Pymolでの可視化
番外編となるが, せっかく構造解析を行ったので, 定番のタンパク質モデル可視化ソフトであるPymolを用いて発表にも使える高画質なタンパク質モデル構造の図を作成する.
Pymolについて詳しくは本記事では述べない. 大阪大学蛋白質研究所 PyMOLの使い方(GUI ver.)のサイトが詳しいので, ぜひともそちらをご参照していただきたい.
実際の作業については, 以下の動画(x5倍速)を参考にしていただきたい.
さいごに
Step.1 ~ Step.4 の計4回で構成されたLysozyme構造のX線結晶構造解析のチュートリアルはここで終了になる. 経験者にとっては当たり前の手順だったかもしれないが, 初めての方にとっては前提知識も含めて学ぶことが多く, 一歩一歩進むのが大変だったと推察する(筆者もすべての作業を言語化して, いかに複雑で高度なことをやっていたのかと驚いた). 大変おつかれ様でした.
本記事を執筆した, 2023/03現在では, タンパク質のX線結晶構造解析は, 人による高度な判断がところどころ入ってしまうため, 完全な自動化が難しいと考えられている. しかし画像生成モデル(Stable Diffusionなど)や大規模言語モデル(GPT3, Chat-GPTなど)に代表されるように, 昨今の急激なAIの進展によって, タンパク質のX線結晶構造解析も, 入力ファイルとして回折画像データセットと対象タンパク質のアミノ酸配列(fasta形式など)を入れ込むだけで精密化された構造が出力される未来はそう遠くはないと思われ, またそうなるべきだと考えている. そのために, 機械学習の技術を有する皆様に, まずは従来の手作業によるタンパク質のX線結晶構造解析の実態を掴んでいただくのが最適なのではと考えたのが, 本記事の執筆のきっかけであった. 初版の執筆が終わってみると, タンパク質X線結晶構造解析を初めて行う学生や研究者の方々にも最適な入門記事になったかなと思う. 多くの方々の助けになれば幸いである.
本チュートリアルに記述した内容は, できるだけ正確性を保っているつもりであるが, ところどころでその分野の専門の方から見たら間違い等があるかもしれない. 本記事をより良くするため, Qiitaのコメント欄などからご意見をいただけると大変幸いである.
また, Step.4ではCCP4のRefmacを用いたが, 実は筆者が得意なのはPhenixを用いた構造精密化である. Step.4番外編としてPhenixを用いた構造精密下の記事も執筆する予定である(おそらく).
最後に, 本記事はところどころにOpenAI/Microsoftが提供しているChatGPT/BingGPTの力をお借りしている. この場をお借りして, それに至るまでの研究者と開発者の血と汗に感謝の意を表したい.
Reference
- タンパク質結晶構造解析入門-ブラックボックスの中身-(4)構造精密化の裏側をのぞいてみよう 精密化
- 2022 DLS-CCP4 Data Collection and Structure Solution Workshop Course Material - Refinement with Refmac
- PDB-101: R-value and R-free
- The phase problem. Garry Taylor, Acta Crystallographica Section D, Volume 59| Part 11| November 2003| Pages 1881-1890