2021年に登場したAlphaFold2をGoogle Colaboratoryで動かす"ColabFold"を使って、コンフォメーション変化をするタイプのタンパク質構造のサンプリングを行う方法を紹介します。
Introduction
ColabFold はGoogle Colaboratoryのサービス上で動くAlphaFold2とその複合体版AlphaFold-Multimerです。2022年5月30日にColabFoldの論文が出ました。この記事の著者である私も3番めに名前がクレジットされています。ColabFoldを使って論文を書いた場合にはぜひ引用してください。
ColabFoldはオリジナルのAlphaFold2の構造推論処理を解析し、Google Colab上で動くようにしただけでなく、それ自体に高速化と高機能化のためのテクニックが含まれています。また、Google ColabはGoogleアカウントがあれば基本的には無料で使用可能なものの、計算の連続使用は90分までに制限されており、さらにその上で1日に何度も使っていると最大12時間ほど利用停止させられてしまうという難点があります。また、無料版Colabで割り当ててもらえるGPUは安物のためにVRAMの値が小さく、アミノ酸で800〜900残基長以上のものは計算できません(それでも十分な場合が多いですが)。そこで、自前でNVIDIA製GPU付きコンピュータを持っている方やスパコンが利用できる方がColabFoldを快適に利用できるために、このlocalcolabfold(というColabFoldのインストーラー)を作成しました。
ここでは、localcolabfoldを使ってその1つであるタンパク質の構造変化のコンフォメーションサンプリングについて説明します。
インストール方法
localColabFoldのインストール方法はここにまとめて書いてあります。Windows10/11(ただしWSL2の環境上での動作), macOS, Linuxのいずれでも動作しますが、NVIDIA製のグラフィックボード、いわゆるNVIDIA製GPUが取り付けられていないPCの場合は動作がとても遅くなります。特にmacOSは今やNVIDIAとの提携を切って久しいため、高速化の恩恵を受けることができません。高速な計算をしたい方はWSL2かLinuxを利用してください。
インストールスクリプトを実行するとcolabfold_batchという名前のディレクトリが生成されます。
(※ 2023年2月中旬のColabFold 1.5.0のリリースに合わせてcolabfold_batchコマンド(メインの実行コマンド)の位置が変わりました)
# Linux用のディレクトリ構成
localcolabfold
│
├── colabfold
│ └── params #Linuxのみ。初回動作時にAlphaFold2のパラメータをこのディレクトリ中にダウンロードしてくる。
├── colabfold-conda # colabfold_batch用のconda環境。lib以下に重要なパッケージがインストールされる。
│ ├── bin
│ | └── colabfold_batch # メインの実行スクリプト。
│ ├── compiler_compat
│ ├── conda-meta
│ ├── data
│ ├── etc
│ ├── include
│ ├── lib
│ ├── ...
├── conda # colabfold_batch用のminiconda環境。
└── update_linux.sh # colabfold_batchのアップデート用スクリプト。
実行のためにはlocalcolabfold/bin/colabfold-conda/binに対して環境変数PATHを追加しておくと良いでしょう。
export PATH="/path/to/localcolabfold/colabfold-conda/bin:$PATH"
これを実行し、which colabfold_batchでlocalcolabfoldへインストール先のPATHが表示されれば成功。この表示は各マシン環境で異なります。
$ which colabfold_batch
/home/moriwaki/Desktop/localcolabfold/colabfold-conda/bin/colabfold_batch
アップデートのやり方
ColabFoldはいまだ開発途中であるため、最新の機能を利用するためにはこのlocalcolabfoldも頻繁にアップデートする必要があります。
アップデートはcolabfold_batchディレクトリの中にあるupdate_linux.shなどのファイルを使って以下のように入力するだけです(最後の.を忘れないでください)。
# Linux/WSL2利用の場合
./update_linux.sh .
# Intel Macを使っている方
./update_intelmac.sh .
# M1 Macを使っている方
./update_M1mac.sh .
localcolabfoldの基本的な使い方
コマンド
colabfold_batch --helpで使い方を確認します。
usage: colabfold_batch [-h] [--stop-at-score STOP_AT_SCORE]
[--stop-at-score-below STOP_AT_SCORE_BELOW]
[--num-recycle NUM_RECYCLE]
[--num-ensemble NUM_ENSEMBLE]
[--random-seed RANDOM_SEED] [--num-models {1,2,3,4,5}]
[--recompile-padding RECOMPILE_PADDING]
....
[--max-msa {512:5120,512:1024,256:512,128:256,64:128,32:64}]
[--zip] [--use-gpu-relax]
[--overwrite-existing-results]
input results
positional arguments:
input Can be one of the following: Directory with fasta/a3m
files, a csv/tsv file, a fasta file or an a3m file
results Directory to write the results to
使い方はcolabfold_batch <options> input resultsで、input, resultsに当たる部分のpositional引数は指定必須です。
inputにはAlphaFold2でおなじみの.fasta形式ファイルだけでなく、.a3mのMSAファイルを指定することで任意のa3mファイルフォーマットを指定可能で、IDと配列をセットにした.csvファイルや、fasta・a3mファイルを含むディレクトリ名を指定することも可能です。
Windows上でWSL2を利用している方のみ以下の環境変数を指定していると計算がうまくいく可能性があります。
export TF_FORCE_UNIFIED_MEMORY="1"
export XLA_PYTHON_CLIENT_MEM_FRACTION="4.0"
export XLA_PYTHON_CLIENT_ALLOCATOR="platform"
export TF_FORCE_GPU_ALLOW_GROWTH="true"
さてcolabfoldの実行コマンドの例は以下の通りです。
$ colabfold_batch \
--amber \
--templates \
--use-gpu-relax \
--num-recycle 3 \
P61823.fasta RNase_monomer
これはP61823.fastaというFASTAフォーマットのファイルを入力としてRNase_monomerディレクトリに結果を出力します。
重要なオプションをいくつか紹介します。
-
--amberをつけると構造最適化を行います。いつもつけておくことが推奨されますが、大量に構造を計算したいときや、後ほどAMBERやGromacsなどで別途構造最適化する方法をユーザーが持っている場合にはこのオプションを付けないというのも手です。 -
--use-gpu-relaxをつけると--amberが有効化されているときにGPUを用いて構造最適化を行います。GPUが存在するマシン上での動作ではこのオプションの利用を強く推奨します。 -
--num-recycle <int>でrecycling回数を指定できます(※詳細はAlphaFold2の論文や仕様書を参照)。AlphaFold2のデフォルト値は3で固定されており、この数字でも十分な精度が出ますが、場合によってはこの数字を20くらいまで大きくすると精度が上がることがあります。ただしその分計算に時間がかかります。 -
--templatesでPDB上に存在する実験構造をテンプレートとして利用します。ただし、colabfoldが参照するPDBデータベースが常に更新されているわけではないため、リリースされたばかりの構造が常に利用できるというわけではありません。その場合、後述の--custom-template-path <directory>を利用することで解決できることがあります。また、現状ではインプットファイルにA3M形式を指定した場合にはテンプレートを考慮した構造推論を利用できないみたいです。 -
--rank {auto,plddt,ptmscore}は構造のランキングの順番をplDDTスコアかpTMscoreかに変更できます。おそらく単量体のときはplDDTを、複合体のときはpTMscoreが自動的に選ばれます。 -
--custom-template-path <directory name>を使うと、そのディレクトリ内に含まれるmmCIFフォーマット(※pdbフォーマットは不可)の構造ファイルのみをテンプレート候補として利用できます。これはコンフォメーション変化をするタイプのタンパク質の構造について、狙ったコンフォメーションをとっている類縁タンパク質がPDBに存在している場合、それに近づけるために利用することがあります。また、AlphaFoldの仕様上model 1,2のみしかテンプレート構造を考慮しないので、これらしか使わないように指定した方が計算時間を節約できます。 -
--trainingをつけると時間がかかる代わりに、Dropoutが適用されて出力結果が変化する……そうです。 -
--random-seed <int>は構造推論時の乱数値を変更します。デフォルトは0で固定されているため、これと入力のMSAが変更されていない限り同じ計算結果が出力されます。 -
--max-msa {512:5120,512:1024,256:512,128:256,64:128,32:64}は構造推論時に利用するMSAの本数を制御します。:の前がAlphaFoldのmax_msa_clustersパラメータで、後者がmax_extra_msaパラメータです。詳しくはAlphaFold論文のSuppl. -
--overwrite-existing-resultsをつけると既存の出力結果を上書きします。
入力ファイルフォーマット
fastaファイル
これは通常のfasta形式と同じです。>で始まる1行目のヘッダーのIDがそのまま出力ファイルのprefixになるため、この行は短い内容にしておくことを推奨します。>以降の配列部分は改行を挟んでもOKです。
>sp|P61823
MALKSLVLLSLLVLVLLLVRVQPSLGKETAAAKFERQHMDSSTSAASSSNYCNQMMKSRN
LTKDRCKPVNTFVHESLADVQAVCSQKNVACKNGQTNCYQSYSTMSITDCRETGSSKYPN
CAYKTTQANKHIIVACEGNPYVPVHFDASV
複合体を予測したい場合は、AlphaFold2と違って:記号で区切りを入れます
>P61823_dimer
MALKSLVLLSLLVLVLLLVRVQPSLGKETAAAKFERQHMDSSTSAASSSNYCNQMMKSRN
LTKDRCKPVNTFVHESLADVQAVCSQKNVACKNGQTNCYQSYSTMSITDCRETGSSKYPN
CAYKTTQANKHIIVACEGNPYVPVHFDASV:
MALKSLVLLSLLVLVLLLVRVQPSLGKETAAAKFERQHMDSSTSAASSSNYCNQMMKSRN
LTKDRCKPVNTFVHESLADVQAVCSQKNVACKNGQTNCYQSYSTMSITDCRETGSSKYPN
CAYKTTQANKHIIVACEGNPYVPVHFDASV
とすればOKです。他のホモ多量体やヘテロ多量体でも入力形式は同じく:で挟むだけです。
また、複数の>のヘッダーと配列情報を1つのfastaファイルに含めて実行することも可能です。
csvファイル
csv形式のフォーマットの例は以下の通りです。idとsequenceをそれぞれ,区切りにします。
id,sequence
5AWL_1,YYDPETGTWY
3G5O_A_3G5O_B,MRILPISTIKGKLNEFVDAVSSTQDQITITKNGAPAAVLVGADEWESLQETLYWLAQPGIRESIAEADADIASGRTYGEDEIRAEFGVPRRPH:MPYTVRFTTTARRDLHKLPPRILAAVVEFAFGDLSREPLRVGKPLRRELAGTFSARRGTYRLLYRIDDEHTTVVILRVDHRADIYRR
やっていることは上のfastaファイルとほぼ同じです。
a3mファイル
すでにcolabfoldやAlphaFoldの計算でa3mフォーマットのMSAファイルを持っている場合は、これを指定することができます。
構造サンプリング
本題の構造サンプリングの方法です。例として、ヒトAcyl-coenzyme A synthetase ACSM2Aの構造サンプリングを行ってみます。これは残基数577で、1つのタンパク質内でアデニル化反応のときとチオエステル化反応のときにコンフォメーションを変化させ、この2つの反応を触媒します。PDBでは2WD9と3C5Eのときにそれぞれのコンフォメーションが見られます。
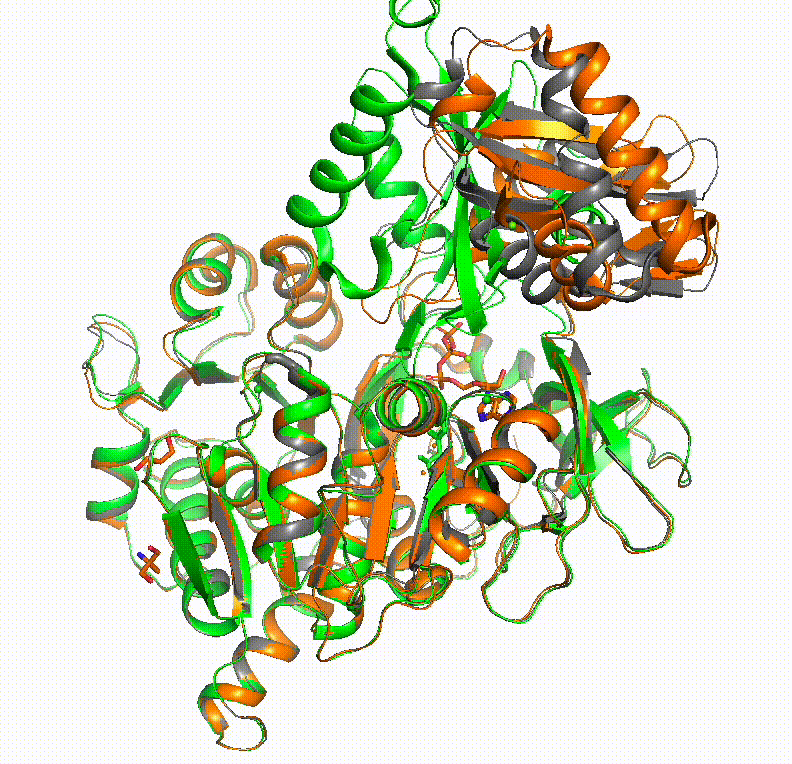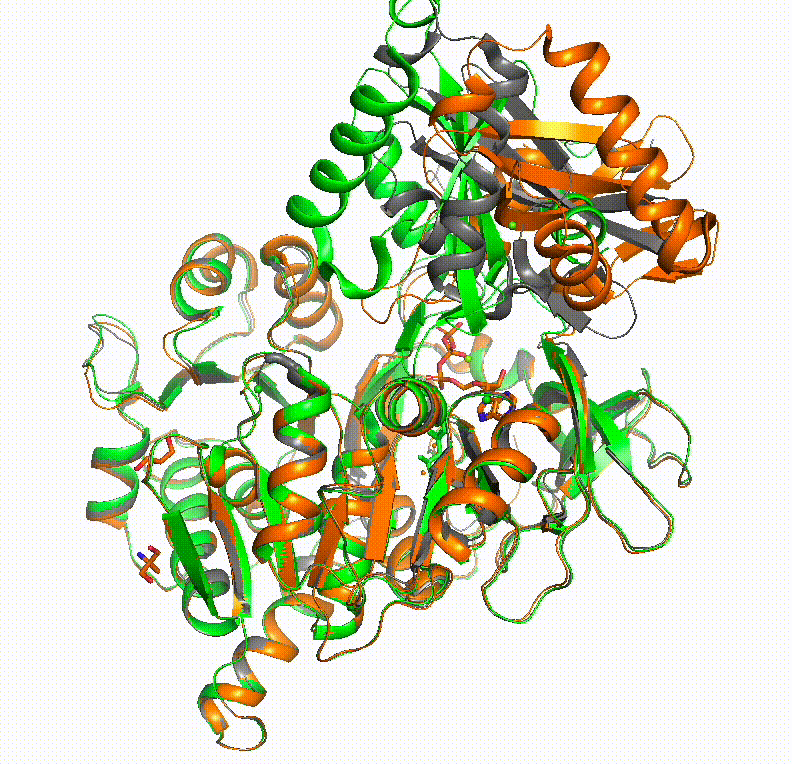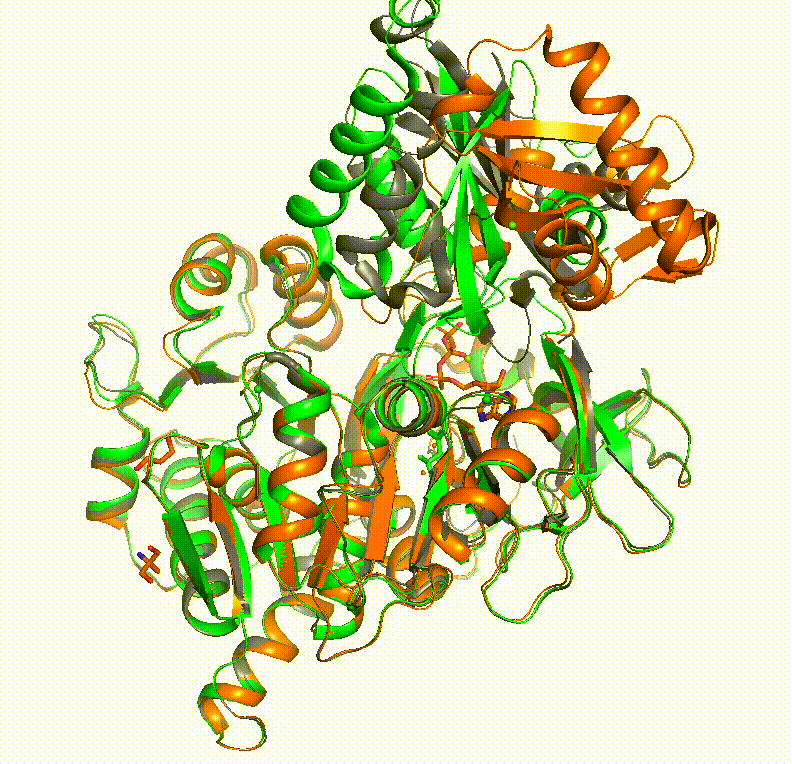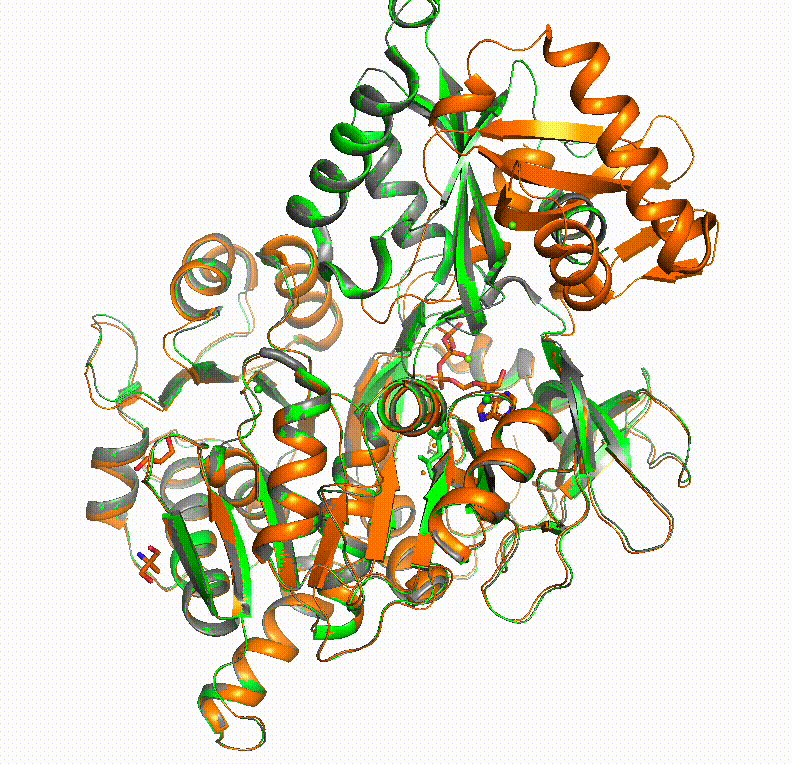
この画像で橙色がアデニル化反応時のコンフォメーション(PDB: 3C5E)、緑色がチオエステル化反応時のコンフォメーション(PDB: 2WD9)です。黒色で構造のmorphingを行っています。使っている配列は以下のQ08AH3.fastaです。
>sp|Q08AH3
MHWLRKVQGLCTLWGTQMSSRTLYINSRQLVSLQWGHQEVPAKFNFASDVLDHWADMEKA
GKRLPSPALWWVNGKGKELMWNFRELSENSQQAANVLSGACGLQRGDRVAVVLPRVPEWW
LVILGCIRAGLIFMPGTIQMKSTDILYRLQMSKAKAIVAGDEVIQEVDTVASECPSLRIK
LLVSEKSCDGWLNFKKLLNEASTTHHCVETGSQEASAIYFTSGTSGLPKMAEHSYSSLGL
KAKMDAGWTGLQASDIMWTISDTGWILNILCSLMEPWALGACTFVHLLPKFDPLVILKTL
SSYPIKSMMGAPIVYRMLLQQDLSSYKFPHLQNCVTVGESLLPETLENWRAQTGLDIRES
YGQTETGLTCMVSKTMKIKPGYMGTAASCYDVQIIDDKGNVLPPGTEGDIGIRVKPIRPI
GIFSGYVDNPDKTAANIRGDFWLLGDRGIKDEDGYFQFMGRANDIINSSGYRIGPSEVEN
ALMEHPAVVETAVISSPDPVRGEVVKAFVVLASQFLSHDPEQLTKELQQHVKSVTAPYKY
PRKIEFVLNLPKTVTGKIQRAKLRDKEWKMSGKARAQ
この構造サンプリングをcolabfoldの--random-seedと--max-msaを組み合わせて実行してみます。実行の仕方はターミナルのbash, zshであれば
#!/bin/bash
INPUTFILE="Q08AH3.fasta"
OUTPUTDIR="ACSM2A"
for RANDOMSEED in `seq 0 9`; do
if test ${RANDOMSEED} -ne 0 ;then
mkdir -p ${OUTPUTDIR}/${RANDOMSEED}
cp -rp 0/*_env ${RANDOMSEED}
fi
colabfold_batch \
--num-recycle 3 \
--num-models 5 \
--model-order 1,2,3,4,5 \
--max-msa 32:64 \
--random-seed ${RANDOMSEED} \
${INPUTFILE} \
${OUTPUTDIR}/${RANDOMSEED}
done
のようにforループさせて実行すればてっとり早いでしょう。これで順番に--random-seedを変化させながら構造推論が実行されます。
ポイントは--max-msa 32:64で利用するMSAの量を制限しているところです。前者は「取得したMSAについて、配列クラスタリングを行うときの中心点とするアミノ酸配列の本数」で後者は「先の配列クラスタリングで使われなかったMSA配列からランダムに追加取得するアミノ酸配列の本数」で、AlphaFold2のデフォルトはそれぞれ512, 5120と設定されています。一般に利用可能なMSAの本数が多いほど構造推論の精度は上がりますが、場合によっては本数が多いとMSAから予測される各残基間の距離予測による補正が強くなりすぎてしまい、1つのコンフォメーションしか生成されなくなります。そこで、これらの値をわざと小さくすることで、この残基関の距離の補正を小さくしています。同じ発想は各種GPCRやtransporterタンパク質についても適用することができ、これらについてもコンフォメーションサンプリングを行うことができます。ただし、適切な--max-msaの値はタンパク質によってまちまちのようで、MSAの数を小さくするとうまく構造が推論できなくなってしまうこととトレードオフの関係になっています。詳しくはこれと同じ手法でGPCR, transporterについてベンチマークを取った論文がありますので、そちらを御覧ください。
これによって、ディレクトリ0から19までが生成されて、その中にそれぞれ5つの予測構造が出力されます。終わったら、これらの構造を結合してムービーにしてみます。構造のまとめ方は色々ありますが、ここではAmberTools22のcpptrajを使って出力されたPDBファイルの残基番号50-460(下半分ドメイン)を重ね合わせてout.pdbという名前で出力してみます。そのために
parm 0/2WD9A_unrelaxed_rank_1_model_2.pdb
trajin ?/2WD9A_unrelaxed_rank_?_model_?.pdb
trajin 1?/2WD9A_unrelaxed_rank_?_model_?.pdb
reference 0/2WD9A_unrelaxed_rank_1_model_2.pdb
autoimage
center :50-460@CA mass origin
rms :50-460@CA
trajout out.pdb
run
というtrajconcat.inファイルを用意します。parm 2WD9A_unrelaxed_rank_1_model_2.pdbの部分は基準とする任意の予測出力構造としています。model_の後の番号はその時によって変化しますので適当に各自の予測結果のファイル名に合わせて修正してください。trajinですべての予測結果pdbファイルを入力とします。
終わったらAmberTools22にPATHが通っている状態でcpptraj -i trajconcat.inを実行し、out.pdbファイルを得ます。PyMOLで見てみると
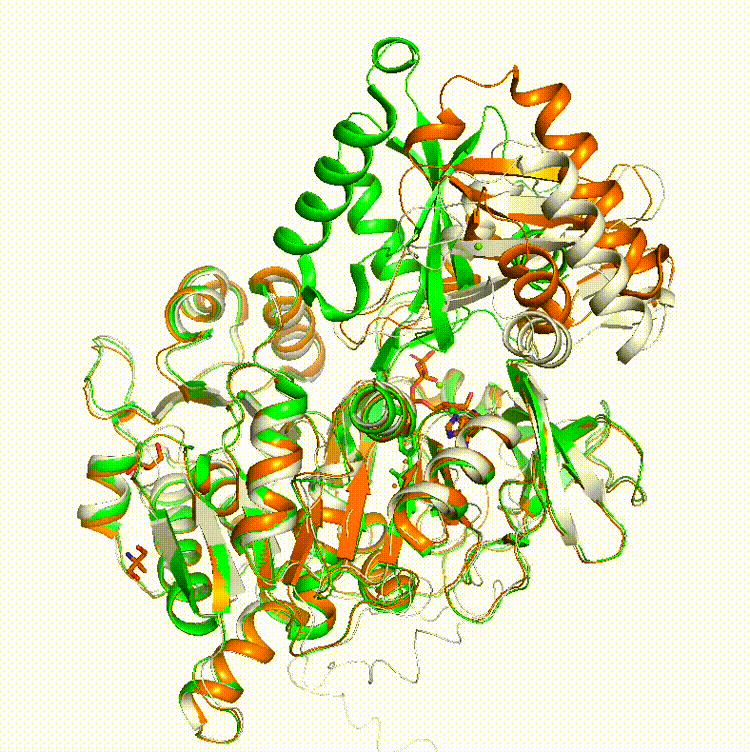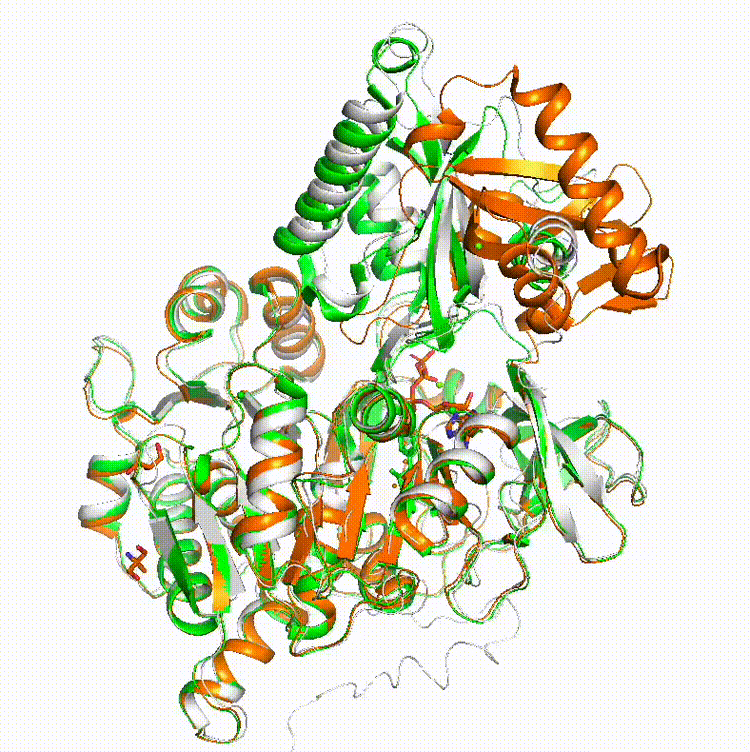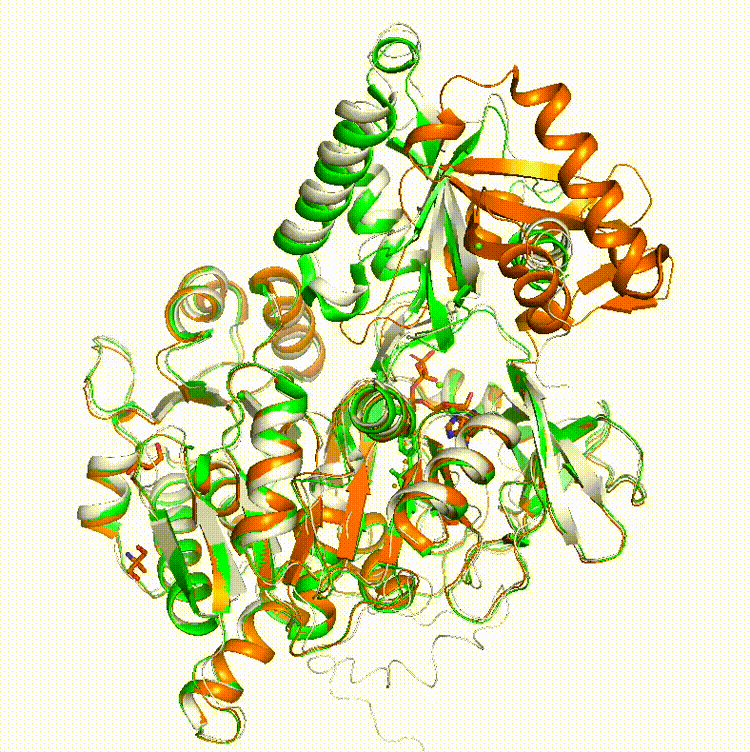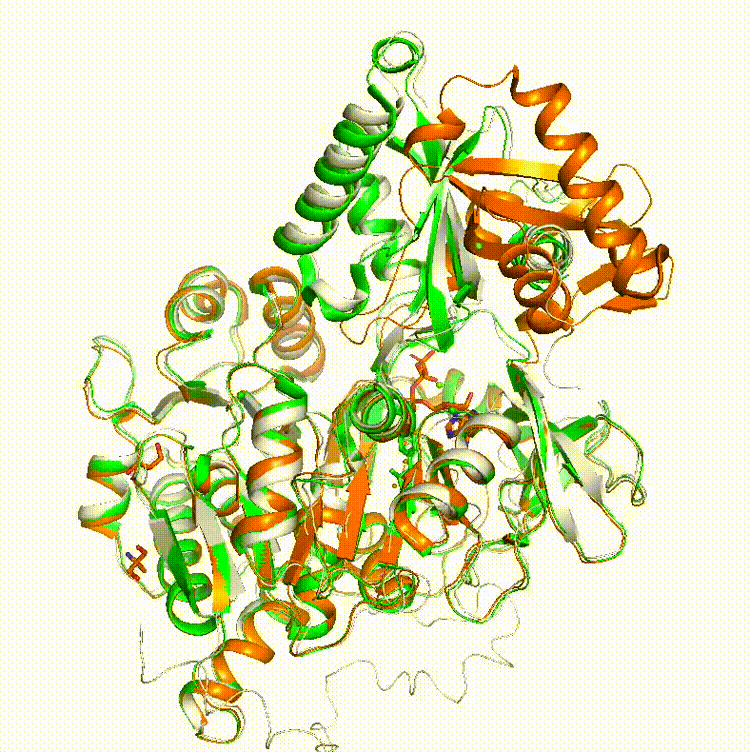
このように、AlphaFoldでは一方のコンフォメーションしか得られなかった構造も、ColabFoldのこの方法でなら複数の安定コンフォメーションをサンプリングする事ができました。あとは適切なところでPyMOLのStateを止めて、その構造をpdb形式で保存すれば1枚1枚に分解できます。
他の例で、ヒトLAT1トランスポーター(PDB: 6IRS_B (open state), 7DSQ_B (closed state) )についても
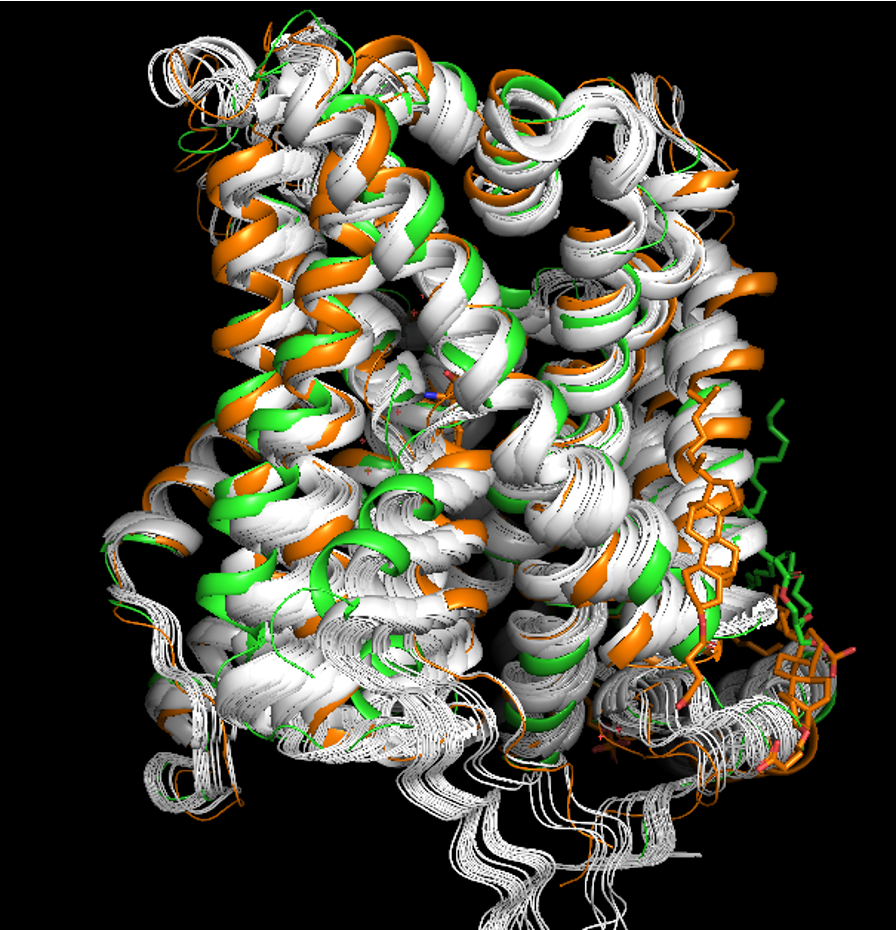
のように、完全にopen状態には至っていませんが、構造サンプリングができています(白色がColabFoldの結果です)。これは先程の論文のFigure 3Aの再現実験です。
References
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S and Steinegger M. ColabFold - Making protein folding accessible to all.
Nature Methods (2022) doi: 10.1038/s41592-022-01488-1 - If you’re using AlphaFold, please also cite:
Jumper et al. "Highly accurate protein structure prediction with AlphaFold."
Nature (2021) doi: 10.1038/s41586-021-03819-2 - If you’re using AlphaFold-multimer, please also cite:
Evans et al. "Protein complex prediction with AlphaFold-Multimer."
BioRxiv (2021) doi: 10.1101/2021.10.04.463034v1