はじめに

画像:© Copyright 2009 - 2019, Aaron R. Quinlan and Neil Kindlon. Last updated on Apr 25, 2020.
Bedtoolsは10年以上も前から活発に開発が続けられている古典的なバイオインフォ用のツールです。ゲノムの位置情報を扱うBEDファイルの演算を行うことができます。BEDファイルはゲノムブラウザに表示することができます。
Githubリポジトリ
https://github.com/arq5x/bedtools2
開発者は Aaron Quinlan さんという方で、ユタ大学でQuinlan Labという研究室を運営しているようです。近年Nim言語やGo言語などで勢力的に高速なツールを開発されているBrent Pedersenさんも、同ラボに所属しています。
この記事ではbedtoolsの初心者が、bedtools公式のリファレンスに加えて、下記の2つの資料をまるごと利用してBedtoolsの使い方を日本語でまとめていきます。
このチートシートではbedの指し示すintervalを「区間」としましたが、かならずしも一般的な用語ではないと思います。必要なら「領域」などと読み替えてください。本文は、ilevantisさんの作成したチートシートをDeepl翻訳、Google翻訳にかけたのち、さらに日本語として意味が通じにくい場所は本家のリファレンスのGoogle翻訳を利用したり書き直すなどして作成しています。画像は公式リファレンスの画像を50%縮小して利用していますが、著作権はすべてBedtoolsの開発チームにあると思います。誤っている部分や改善点がございましたら、プルリクエストやコメント欄にコメントください。
コマンドの全体像
一般的な操作
| サブコマンド | 説明 |
|---|---|
| flank | 既存の区間に隣接した新しい区間を作成します。 |
| slop | 区間のサイズを調整します。 |
| shift | 区間の位置を調整します。 |
| subtract | 2つの区間の重複を削除します。 |
| complement | 指定した区間以外の領域を区間として抽出します。 |
| closest | 最も近い区間を検索します。 |
| intersect | 重複した区間を抽出します。 |
| window | 区間を含むウィンドウで重複する区間を検索します。 |
| cluster | 重なっている区間や近い区間をクラスタ化します。(マージはしません。) |
| merge | 重なっている/近くにある区間を 1つの区間に結合します。 |
| map | 重複している区間の集合に関数を適用します。 |
| groupby | 列でグループ化し、他の列をまとめる。 |
ファイル形式
| サブコマンド | 説明 |
|---|---|
| getfasta | BEDで指定した区間の配列を出力します。 |
| maskfasta | BEDで指定した区間以外をNにしたfastaを返す。 |
| sort | 区間をソートします。 |
| bed12tobed6 | BED12 を BED6 に分割します。 |
| bamtofastq | BAM 形式を Fastq 形式に変換します。 |
| bamtobed | BAM 形式を BED 形式に変換します。 |
| bedpetobam | BEDPE 形式を BAM 形式に変換します。 |
| bedtobam | BED 形式を BAM 形式に変換します。 |
統計
| サブコマンド | 説明 |
|---|---|
| jaccard | ジャカード係数を計算します。 |
| random | ランダムな区間を生成します。 |
| reldist | 2つのファイルの相対的な距離の分布を計算します。 |
| shuffle | 区間をランダムに再配置します。 |
| makewindows | Makes adjacent or sliding windows across a genome or BED file. |
| nuc | 区間内のヌクレオチド含有量を計測します。 |
カバレッジ
| サブコマンド | 説明 |
|---|---|
| annotate | 1つのファイルに他のファイルとのカバレッジや重複の数で注釈をつける |
| coverage | 定義された間隔のカバレッジを計算します。 |
| genomecov | ゲノム全体のカバレッジを計算します。 |
| multicov | 特定の間隔で複数のBAMからのカバレッジを計算します。 |
| unionbedg | 複数のBEDGRAPHファイルのカバレッジ間隔を結合します。 |
共通オプション
| オプション | 説明 |
|---|---|
| -s | 同じストランドでAと○○するBのヒットのみを報告します。 |
| -S | 反対のストランドでAと○○するBのヒットのみを報告します。 |
| -f | Aの分数として必要な最小オーバーラップ。デフォルトは1E-9(つまり1bp)です。 |
| -F | Bの分数として必要な最小のオーバーラップ。デフォルトは1E-9(つまり、1bp)です。 |
| -r | オーバーラップの割合がAとBの相互的であることを要求します。つまり、-fが0.90で、-rが使用されている場合、BはAの少なくとも90%とオーバーラップし、Aも少なくともBの90%とオーバーラップする必要があります。 |
| -e | AまたはBのいずれかを満たす必要があります。つまり、-eを-f 0.90および-F 0.10と一緒に使用する場合は、Aの90%がカバーされるか、Bの10%がカバーされる必要があります。-eがなければ、両方が満たされる必要があります。 |
| -split | BAMまたはBED12のエントリを分割したものをBEDの区間として扱う。 |
| -abam | A は BAMファイル。 |
一般的な操作
flank
既存の区間に隣接した新しい区間を作成します。 (flank Docs)
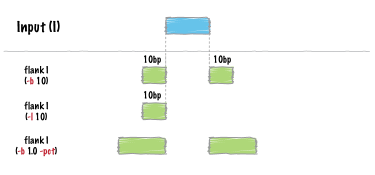
IN ▓▓▓▓▓ ▓▓▓
Flank ██ ██ ██ ██
Slop █████████ ███████
bedtools flank [オプション] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
| オプション | 説明 |
|---|---|
| -b, -l, -r | 両側に、左側に、または右側に、x bpずつ領域をフランク/拡張します。 |
| -s | ストランドに基づいて -l と -r を定義します。 |
| -pct | l と -r を、特徴(feature)の長さの割合として定義します。 |
slop
区間の大きさを調整します。 (slop Docs)
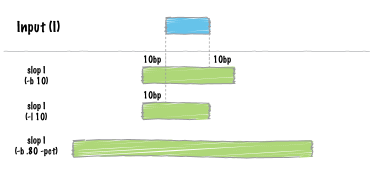
bedtools slop [オプション] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]`
| オプション | 説明 |
|---|---|
| -b, -l, -r | 両側に、左側に、または右側に、x bpずつ領域をフランク/拡張します。 |
| -s | ストランドに基づいて -l と -r を定義します。 |
| -pct | l と -r は、特徴(feature)の長さの割合で定義します。 |
shift
染色体のへりを尊重しながら、間隔の位置を調整する (Docs).
IN ██ ██ ████
OUT ██ ██ ████
bedtools shift [オプション] -i <BED/GFF/VCF> -g <GENOME> [-s or (-m and -p)]
| オプション | 説明 |
|---|---|
| -s | シフトする塩基の数(BPs)。 |
| -m, -p | マイナス鎖 または プラス鎖 においてシフトする塩基の数 |
| -pct | s、-m、-pを特徴の(feature)長さの比率として定義します。 |
subtract
2つのファイルの重なりに基づいて区間を削除します。 (Docs)
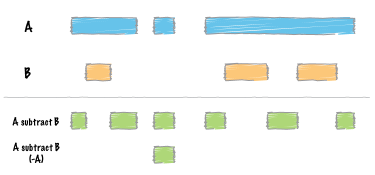
A ▓▓▓▓▓▓▓▓▓▓ ▓▓▓ ▓▓▓▓▓▓
B ▓▓▓▓ ▓▓▓▓▓▓▓
A sub B ██ ████ ███ ███
bedtools subtract [オプション] -a <BED/GFF/VCF> -b <BED/GFF/VCF>
| オプション | 説明 |
|---|---|
| -A | 重複している場合は、特徴(feature)全体を削除します。 |
| 共通 | strandedness: -s, -S; overlap: -f, -F; overlap mode: -r, -e |
complement
BEDファイルで表されない領域を抽出します。(Docs)
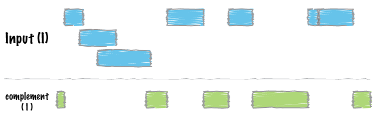
IN ▓▓▓▓▓ ▓▓▓ ▓▓▓▓▓▓
▓▓▓▓ ▓▓▓
OUT █████ █████ ██
bedtools complement -i <BED/GFF/VCF> -g <GENOME>
closest
最も近い区間を見つけます。 (Docs)

A █████ ✓
B ████ ███
bedtools closest [オプション] -a <FILE> -b <FILE1, FILE2, ..., FILEN>
| オプション | 説明 |
|---|---|
| -d | Aから最も近い特徴までの距離も報告します。 |
| -k | k個の最も近いヒットを報告します。デフォルト:1。 |
| -io | Aと重複するBの特徴を無視します。 |
| -iu, -id | Aの特徴の上流または下流にあるBの機能を無視します。 |
| 共通 | strandedness: -s, -S |
intersect
重複する間隔をさまざまな方法で見つけます。 (Docs)
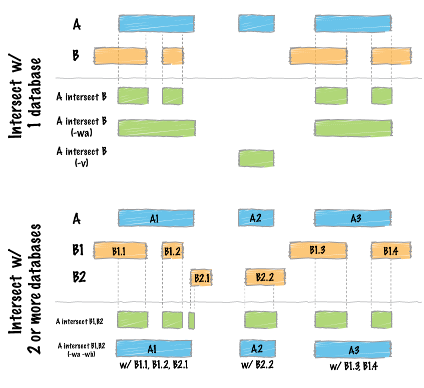
A ██████████
B ▓▓▓▓ ▓▓ ▓▓▓
A int B ▓▓ ▓▓
$ bedtools intersect [オプション] -a <BAM/BED/GFF/VCF> -b <FILE1, FILE2, ..., FILEN>
| オプション | 説明 |
|---|---|
| -wa, -wb | 重複ごとに、Aの元のエントリ/ Bの元のエントリをそれぞれ書き込みます。 |
| -loj | 「左外部結合」を実行します。つまり、Aの各フィーチャについて、Bと重複しています。重複が見つからない場合は、BのNULLフィーチャをレポートします。 |
| -wao | 元のAエントリとBエントリに加えて、2つのフィーチャ間のオーバーラップのベースペアの数を書き込みます。オーバーラップのあるAフィーチャーのみが報告されます。-fおよび-rによって制限されます。 |
| -u | Only report each overlapping A feature once. |
| -c | Aの各エントリについて、重複するBの特徴の数を報告します。 |
| -v | Bと重複しないAの特徴を報告します |
| 共通 | strandedness: -s, -S; overlap: -f, -F; overlap mode: -r, -e; bam/bed12: -abam, -split |
window
区間を中心としたウィンドウ内で重なり合う区間を見つけます。 (Docs)
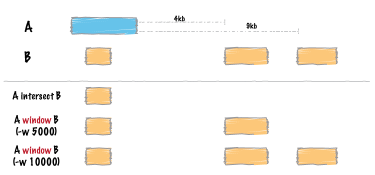
A ┌────█████────┐
B ▓▓▓▓ ▓▓▓ ▓▓▓
A win B ▓▓▓▓ ▓▓▓
bedtools window [オプション] [-a|-abam] -b <BED/GFF/VCF>
| オプション | 説明 |
|---|---|
| -w, -l, -r | 両方向、上流、下流のオーバーラップウィンドウのフランク長。 |
| -sw | ストランドに基づいて-lおよび-rを定義します。 |
| -u | Only report each overlapping A feature once. |
| -c | Aの各エントリについて、重複するBの特徴の数を報告します。 |
| -v | Bと重複しないAの特徴のみ報告します |
| 共通 | strandedness: -sm, -Sm; bam: -abam |
cluster
重なった、または近傍にある区間をまとめる(マージはしない)(Docs)
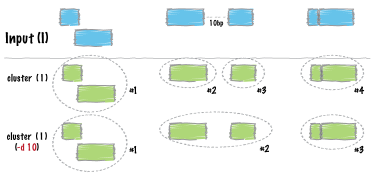
BED ████ █████ ███
clustID └─#1─┘ └────#2────┘
bedtools cluster [オプション] -i <BED/GFF/VCF>
| オプション | 説明 |
|---|---|
| -d | クラスタ内の特徴(feature)間の最大距離。 |
| 共通 | strandedness: -s, -S |
集計ツール
merge, groupby, map では、-c で指定されたカラム/列に以下の*集計関数(-oで指定)を適用することができます。
sum, count, count_distinct, min, max, mean, median, mode, antimode, stdev, sstdev, collapse, distinct, first, last
※その他の機能もあります。
merge
重なり合う/近接している区間を1つの区間に結合します。 (Docs)
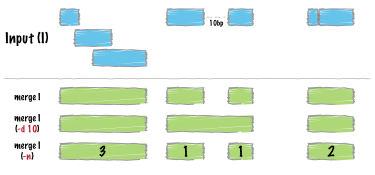
IN ▓▓▓ ▓ ▓▓··d··▓▓▓
▓▓▓▓ ▓▓
OUT ██████ ███ ██████████
$ bedtools merge [オプション] -i <BED/GFF/VCF/BAM>
| オプション | 説明 |
|---|---|
| -s | Require same strandedness. |
| -S | Force merge for one specific strand only. Options: <`+`/`-`>. |
| -d | マージする特徴(feature)間の最大距離。 |
| 共通 | aggregation: -o, -c; |
map
重複する区間を列とみなして関数を適用します。(Docs)
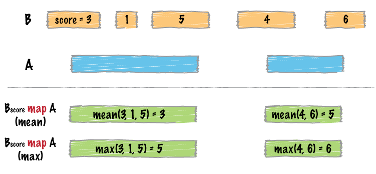
score = 3 1 5 4 6
B ▓▓▓ ▓ ▓▓▓▓▓ ▓▓▓▓▓▓ ▓▓▓▓
A ██████████ ███████
B map(mean) A ██████████ mean(3,1,5)=5 ███████ mean(4,6)=5
bedtools map [オプション] -a <BED/GFF/VCF> -b <BED/GFF/VCF>
| オプション | 説明 |
|---|---|
| 共通 | aggregation: -o, -c; strandedness: -s, -S; overlap: -f, -F; overlap mode: -r, -e; bed12: -split |
groupby
列でグループ化し、他の列を要約します (~ SQL "groupBy"). (Docs)
bedtools groupby [オプション] -i <BED> -g <groupby columns> -c <op. column> -o <operation>
| オプション | 説明 |
|---|---|
| common | aggregation: -o, -c |
ファイル形式
BED形式
| 列 | 例 | 定義 |
|---|---|---|
| chrom | Sc112.1 | <STR> chromosome/scaffold の名前 |
| start | 2134 | <INT> 特徴(feature)の開始位置 |
| end | 2565 | <INT> 特徴(feature)の終了位置 |
| name | gene123 | <STR> 特徴(feature)の名前 |
| score | 544 | <NUM> 特徴(feature)のスコア (ビットスコアなど) |
| strand | + | <+/-/.> どちらの鎖に特徴(feature)が位置するか |
| thickStart | 2235 | |
| thickEnd | 2489 | |
| itemRgb | 255,0,0 | |
| blockCount | 2 | <INT> 特徴(feature)のブロックの数(Exonの数) |
| blockSizes | 150,80 | <INT>,<INT>,... ブロックのサイズのリスト |
| blockStarts | 0,2333 | <INT>,<INT>,... 特徴(feature)の開始位置を基準としたブロックの開始位置のリスト |
こちらも参照 → BEDフォーマット macでインフォマティクス
GFF と BED のindexの違い
こちらも参照 → BEDファイルの座標の注意点
GFF ┌─1 2 3─┐ 4 ...
G---A---T C ...
BED └─0 1 2 └─3 ...
| . | gff -> bed | bed -> gff |
|---|---|---|
| new_start = | gff_start - 1 | bed_start + 1 |
| new_end = | gff_end | bed_end |
getfasta
区間を使用して、FASTAファイルからシーケンスを抽出します。(Docs)

FASTA ACTGATCATGATACATGATACCATTAGGATACAATA
BED ████ █████ ████
OUTFA ATCA TGATA GGAT
bedtools getfasta [オプション] -fi <input FASTA> -bed <BED/GFF/VCF>
| オプション | 説明 |
|---|---|
| -name | FASTAヘッダーに、BEDファイルの「name」列を使用します。 |
| -s | マイナス鎖を逆補完する機能 デフォルト:ストランド情報は無視されます。 |
| -split | BED12入力が与えられた場合、BEDブロック(例:エクソン)からのシーケンスを連結します。 |
maskfasta
区間を使用して、FASTAファイルのシーケンスをマスクします。 (Docs)
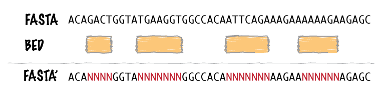
FASTA ACTGATCATGATACATGATACCATTAGGATACAATA
BED ████ █████ ████
FASTA' ACTGATNNNNATACATGNNNNNATTAGGNNNNAATA
bedtools maskfasta [オプション] -fi <input FASTA> -bed <BED/GFF/VCF> -fo <output FASTA>
| オプション | 説明 |
|---|---|
| -soft | ソフトマスク (Nではなく小文字に変換する) |
| -mc | マスキング文字を指定します。 |
sort
区間を並び替えます。 (Docs)
bedtools sort [オプション] -i <BED/GFF/VCF>
| オプション | 説明 |
|---|---|
| -sizeA | サイズ(asc:昇順)で並べ替えます。 |
| -sizeD | サイズ (desc:降順) で並び替えます。 |
| -chrThenSizeA | 染色体順 (asc:昇順 サイズ (asc:昇順)。 |
| -chrThenSizeD | 染色体順 (asc:昇順) サイズ (desc:降順)。 |
| -chrThenScoreA | 染色体順 (asc:昇順) スコア (asc:昇順)。 |
| -chrThenScoreD | 染色体順 (asc:昇順) スコア (desc:降順)。 |
統計
jaccard
ジャカード係数を計算します。(Docs)
Jaccard係数: X または Y に含まれている要素のうち X にも Y にも含まれている要素の割合
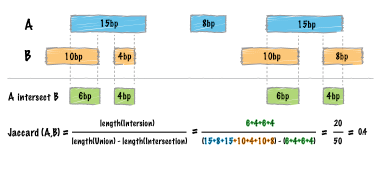
A ███████████ 15bp
B ▓▓▓▓ 10bp ▓▓ 4bp ▓▓▓ 8bp
A int B ▓▓ 6bp ▓▓ 4bp
Jaccard(A,B) (6+4)/((15+10+4+8)-(6+4)) = 0.37
bedtools jaccard [オプション] -a <BED/GFF/VCF> -b <BED/GFF/VCF>
| オプション | 説明 |
|---|---|
| 共通 | strandedness: -s, -S; overlap: -f, -F; overlap mode: -r, -e; bed12: -split |
random
ランダムな区間を生成します。(Docs)

bedtools random [オプション] -g <GENOME>
| オプション | 説明 |
|---|---|
| -l | 生成する区間の長さ。デフォルト:100 |
| -n | 生成する区間の数。デフォルト:1,000,000 |
| -seed | 整数でシードを指定します。 |
reldist
2つのファイルの相対的な距離の分布を計算します。(Docs)
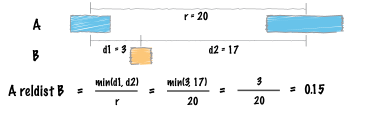
───────r──────
A ▓▓▓▓▓▓ ▓▓▓▓
B ███
───d1─── ──d2──
reldist = min(d1,d2)/r
bedtools reldist [オプション] -a <BED/GFF/VCF> -b <BED/GFF/VCF>
| オプション | 説明 |
|---|---|
| -detail | 要約の代わりに、Aの各領域の相対距離を報告します。 |
shuffle
区間をランダムに再配置します。(Docs)
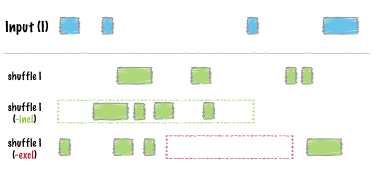
bedtools shuffle [オプション] -i <BED/GFF/VCF> -g <GENOME>
| オプション | 説明 |
|---|---|
| -excl | 特徴を配置してはならない座標のBEDファイル。 |
| -incl | 特徴を再配置する必要がある座標のBEDファイル。 |
| -chrom | 同じ染色体上に特徴(feature)を保持する。 |
| -chromFirst | ゲノム全体(デフォルト)からランダムに位置を選択する代わりに、最初にランダムに色を選択し、次にその色のランダムな開始座標を選択します。これにより、特徴が染色体サイズの関数として分布するのとは対照的に、特徴が染色体間で均一に分布します。 |
| -noOverlapping | 再配置された区間が重ならないようにする。 |
makewindows
Makes adjacent or sliding windows across a genome or BED file.
bedtools makewindows [オプション] [-g <GENOME>|-b <BED>] [-w <window size> | -n <n windows>]
| オプション | 説明 |
|---|---|
| -s | Number of bases to step before creating a new window. Default: equal to -w
|
Coverage
annotate
1つのBED / VCF / GFFファイルに、他の複数のBED / VCF / GFFファイルから観測されたカバレッジと重複の数を注釈します。 (Docs)
$ bedtools annotate -i variants.bed -files genes.bed conserve.bed known_var.bed
chr1 100 200 nasty 1 - 0.500000 1.000000 0.300000
chr2 500 1000 ugly 2 + 0.000000 0.600000 1.000000
bedtools annotate [オプション] -i <BED/GFF/VCF> -files FILE1 FILE2 FILE3 ... FILEn
| オプション | 説明 |
|---|---|
| -counts | -iと重複する各ファイルの特徴(feature)の数を報告します。デフォルトの動作では、各ファイルがカバーする-iの割合を報告します。 |
| -both | 各ファイルについてカウントと割合をレポートします。 |
| 共通 | strandedness: -s, -S. |
coverage
指定した区間のカバレッジの深さと幅の両方を計算します。 (Docs)
BED FILE A ▓▓▓▓▓▓▓▓▓▓▓▓▓▓▓ ▓▓▓▓▓▓▓▓▓▓▓▓▓▓▓ ▓▓▓▓▓▓
BED File B ████ ████ ██ █████████
████████
Result [ N=3, 10/15 ] [ N=1, 2/15 ] [N=1,6/6]
bedtools coverage [オプション] -a <BAM/BED/GFF/VCF> -b <FILE1, FILE2, ..., FILEN>
| オプション | 説明 |
|---|---|
| -d | Aの特徴(feature)の各位置の深度を報告します。 |
| 共通 | strandedness: -s, -S; overlap: -f, -F; overlap mode: -r, -e; bam/bed12: -split,-abam |
その他のサブコマンド
-
expand
-
fisher
- 2セットの間隔の間のオーバーラップの量が、それらのカバレッジとゲノムのサイズを考慮して、予想よりも多いかどうか
-
genomecov
-
igv
- IGVスナップショットバッチスクリプトを作成します。
-
links
- UCSCの場所へのリンクのHTMLページを作成します。
-
multiinter
-
overlap
- 指定したフィーチャ間のオーバーラップの量
-
pairtobed
-
pairtopair
-
tag
おわりに
私の理解が浅いので、この記事はここまでです。バイオインフォでは、日本語訳の定番が決まっていない単語がたくさんあるような気がしています。英語は英語のママ理解すればいいじゃないかという人もいるかも知れませんが、日本語の情報が増えれば、私達もネイティブに考えやすくなると思います。もっとbedtoolsに対する理解が深まったら加筆・修正をかけたいと思います。