よく使うbedファイル。ここにきて全く知らなかったbedファイルの特殊性について、大反省するためにここに書こう。
現在私は、1base毎の計算が必要な解析をしている (SNVなど)。genomic coordinatesを扱う時にとても便利なbed fileは、sam,bam,vcfなどいろいろなファイル形式からおこせるシンプルで便利なfile formatだ。
ところが、ここで大きな落とし穴があった。問題はUCSCのFAQに記載がある。
The first three required BED fields are:
chrom - The name of the chromosome (e.g. chr3, chrY, chr2_random) or scaffold (e.g. scaffold10671).
chromStart - The starting position of the feature in the chromosome or scaffold. The first base in a chromosome is numbered 0.
chromEnd - The ending position of the feature in the chromosome or scaffold. The chromEnd base is not included in the display of the feature.
For example, the first 100 bases of a chromosome are defined as chromStart=0, chromEnd=100, and span the bases numbered 0-99.
一体何を書いているのかというと、
「bedファイルに記載している染色体の位置座標は0-based startである。」
ということだ。なんだそんなことかと思うかもしれない。だが待ってほしい。これは本当に重要なことで、例えば染色体1番の1-10番までの塩基を示すには、
chr1 0 10
という記載が必要になってくる。座標の長さを計算するときは単純な引き算で済むので、この表記は楽ではあるが何よりも1baseが重要な研究になってくると1baseのズレが命取りになってくる。もう少し分りやすく、ブラウザ上での表示をみてみよう。こういう時に、UCSC genome browserのcustom trackは便利だ。
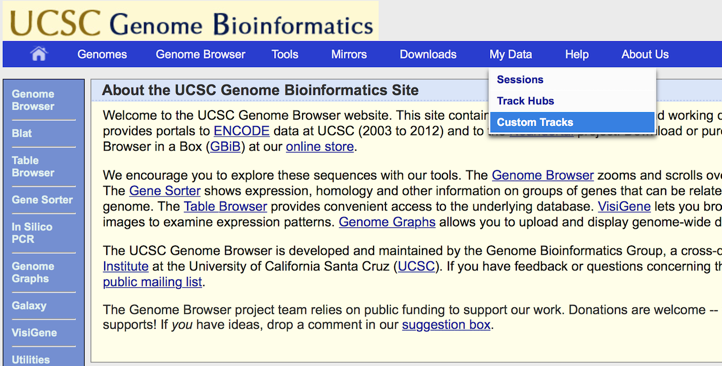
次のページにコピペでデータが入力できるようになっている。UCSCで読み込むデータを作る前には簡単なテストデータを作った上でここで確認を行ったりすることによく使っている。
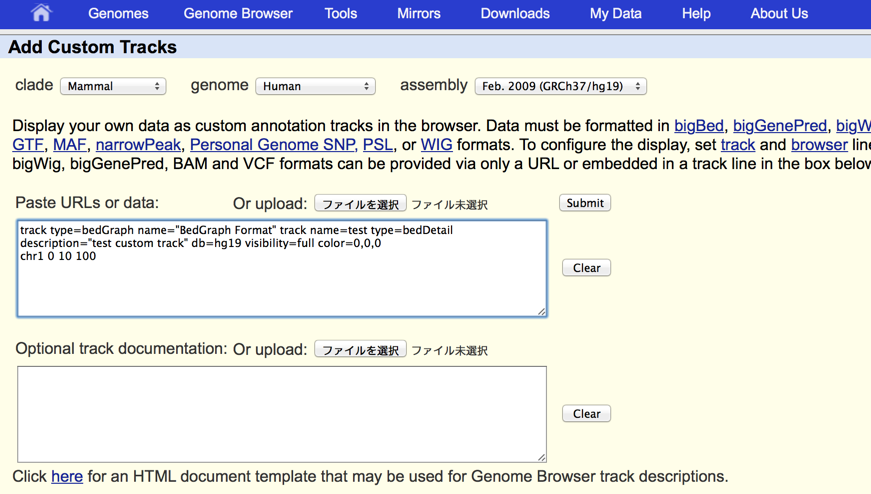
例では、manualからテキトウにbedGraph形式のheaderを追加し、領域が分りやすいように100という値も追加した。
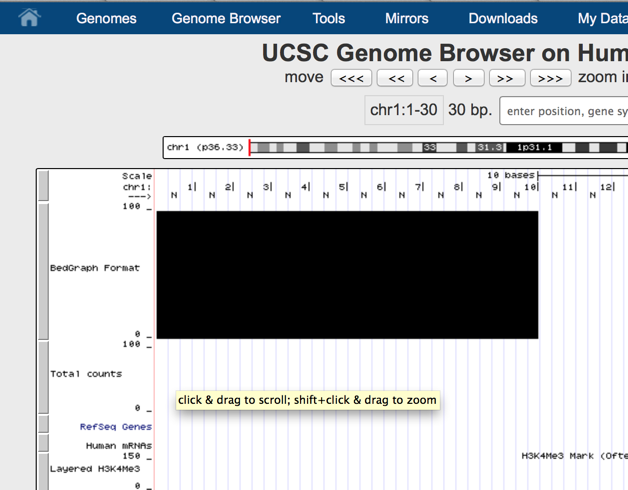
実行結果。Nが塩基に相当する。hg19の染色体1番の開始塩基はまだ解読されていないので、Nになる。黒いバーのところがbedファイルで指定した領域。N10個分が黒いバーに入っている。
1から10までの番号の間に塩基配列Nが入っていることがわかる。このようにbedファイルは、染色体の塩基の座標を示しているのではなく、塩基の間に座標をふったものと考える方がいいだろう。確かに、bedファイルは座標情報を記載しているだけであり、塩基情報を捨ててしまっているので当然といってもいいのかもしれないが、他のデータから、bedファイルへ情報を移すときには注意が必要だ。
例えば、MACSでpeak callを行った時のsummit情報(peakの頂点情報)を扱う時やCAGEデータでは、その1塩基の情報が0-startedか、1-startedであるかをしっかり見極めないといけない。おそらく、最初からbed形式のデータを出力する場合は、0-started、その他のデータの場合は1塩基に相当する座標が必要になるので、1-started(塩基に座標がふってある)と考えるのが妥当だろう。
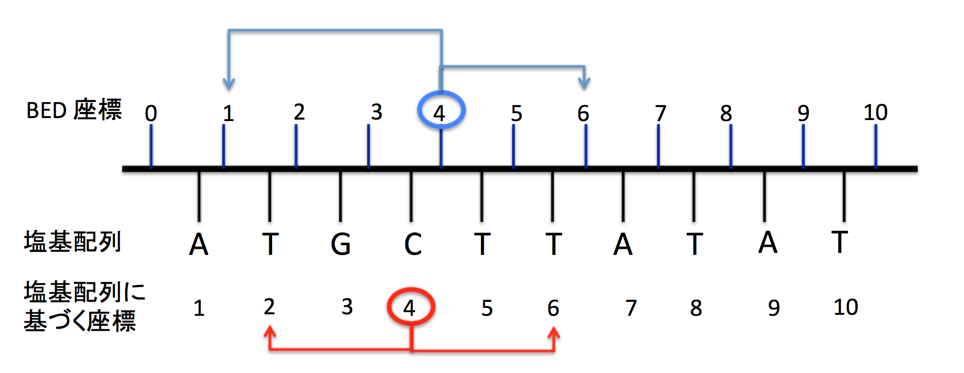
例えば、上の例をもとに4 番目の塩基をsummitとしたmotif解析を考える場合、
4番塩基から上流下流の2塩基をbedファイルからfastaファイルを生成し、取得する事を考える。bedファイルから塩基配列情報に変換する場合に最終的に「TGCTT」を取得できるように変換しなくてはならないが、4番塩基はbed座標では、3-4間に存在する塩基なので、bedファイルは、
chr1:1 6
となる。fasta, fastqファイルなどを直接弄る場合はその必要はないだろう。2番から6番までの塩基を取得すればよい。
IGVとUCSC genome browserの挙動の違い。
上記を踏まえてさらに進むと、UCSC genome browserではbedGraphやbigWigファイルの座標指定に、
chr1 100 100
のようなstart,endの座標を一致させた場合は可視化できない。
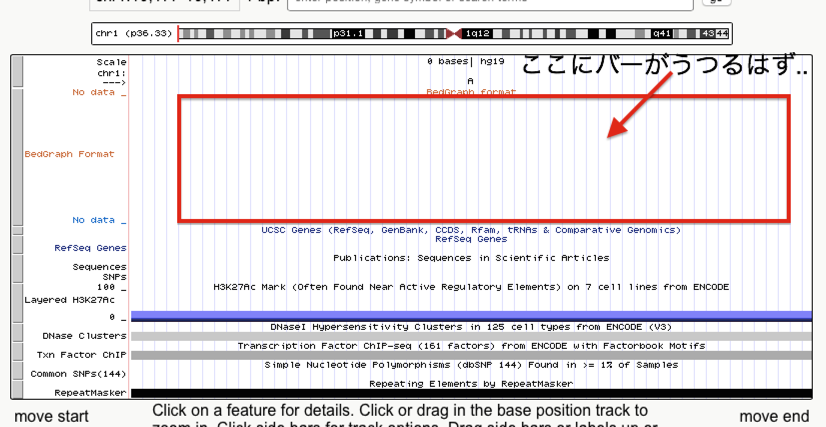
これは、そもそもbedファイルは塩基配列の座標ではないからだ。すなわち、start,endの座標を同じにすることなどもってのほかであり、正しくは、
chr1 99 100
という記述が正しくなる。
ところがIGVでは、
chr1 100 100
のような1塩基座標が指定できる。
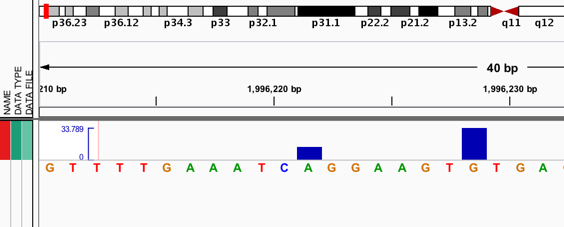
IGVはVCFやGFFファイルの読み込みにも対応しているのでそういう仕様なのかもしれないが、個人的にはIGVの仕様の方が直感的だ。でも、計算は0-startedの方がやりやすいのは事実だ。