概要
本稿では、菌叢解析ソフト Qiime2(2019.7 ver.)を用いて、細菌の系統分類マーカーである 16S rRNA 遺伝子(16S rDNA)のアンプリコン(PCR増幅産物)から、微生物群集構造を解析する方法**(16S アンプリコン解析)**を紹介する。本稿で紹介する解析フローやコマンドは一部を除いて別バージョンの Qiime2 でも共通である。(2020.2 版、2020.6 版、2020.8 版、2021.2 版では確認済。)
なお、系統分類マーカーだけでなくゲノム全体を対象にしたショットガンメタゲノム解析は本稿の対象ではない。
菌叢解析手順
以下に、16S アンプリコンを用いた菌叢解析手順を 4 段階でまとめた。アンプリコン解析で得られるデータセットは、試料から抽出されたサンプルの read 数であって、試料に含まれる細菌の絶対的な存在量ではない。また、シーケンサーの性能や PCR の精度によって、サンプルの合計 read 数は変わる。したがって、サンプルは細菌の組成に基づいて比較されるべきである。
- 試料からサンプルとして DNA を抽出する。
- DNA をシーケンシングする。
- 「塩基配列」と「各塩基配列の read 数」を得る。
- read 数から、各サンプルの細菌組成を比較する。
Qiime2 による解析の工程
16S アンプリコン解析において、Qiime2 では以下の工程で解析を実施する。

まずアンプリコン配列は、シーケンシング時に PCR で生じたエラーを修正される。ここで「塩基配列」と「各塩基配列の read 数」が得られる。これを、塩基配列のまま比較することも可能だが、データベース上の細菌に照合してから比較したり、細菌がもつ酵素の量を予測して比較したりすることもできる。比較は、**組成データ解析(Compositional data analysis:CoDA)**によって行われる。
各解析の概要
-
Taxonomy analysis
16S rDNA のアンプリコンシーケンスをデータベース上の細菌に照合させる分析。どのような細菌が存在するのかを明らかにする。 -
PICRUSt2 analysis
群集中の細菌がコードしている酵素の存在量を予測する解析。専用ツールの PICRUSt2 はデフォルトの Qiime2 にはインストールされないため、 別個にプラグインする必要がある。ただし、最新版の Qiime2 には対応していないことがあるため、本稿では 2019.7 版をインストールしている。 -
Differential analysis
群間で、特徴量(細菌や酵素)の存在量を比較する多変量解析。ANCOM、Balance、ALDEx2 などがある。存在量に差がある特徴量の索敵を目的とする。 -
Diversity analysis
群間で、特徴量(細菌や酵素)の多様性を比較する多変量解析。α-多様性とβ-多様性がある。
テストデータ
本稿では IBD multi'omics database (IBDMDB) に公開されている腸生検の 16S rDNA アンプリコンシーケンスをテストデータとして採用し、クローン病(CD)患者、潰瘍性大腸炎(UC)患者、非炎症性腸疾患(nonIBD)被験者における腸内細菌叢および菌叢代謝を比較した。このデータセットには合計 178 サンプルが登録されている。
各データは、File Name(206172.tarなど)をクリックするとダウンロードできる。この記事を参考に動作確認をしてみたい場合は、いくつかのサンプルをダウンロードしてみるとよい。
ダウンロードした tar ファイルを展開すると .bz2 に圧縮されている 2つの FASTQ ファイル(拡張子.fq)が得られる。解析のため、.bz2 も展開しておこう。
末尾が 「.1.fq」 のファイルは forward reads で、末尾が「.2.fq」のファイルは reverse reads である。
FASTQ 形式
テストデータには、シーケンシングによって得られた read の塩基配列と品質情報が FASTQ 形式 で記載されている。各 read の情報は以下の 4 行でまとめられている。
- 1行目:ID
- 2行目:塩基配列
- 4行目:品質情報
@M70287:115:000000000-B59YV:1:1101:18877:1845 1:N:0:
TACGGAGGATCCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGAGCGTAGGTGGACTGGTAAGTCAGTTGTGAAAGTTTGCGGCTCAACCGTAAAATTGCAGTTGATACTGTCAGTCTTGAGTACAGTAGAGGTGGGCGGAATTCGTGGTGTAGCGGTGAAATGCTTAGATATCACGAAGAACTCCGATTGCGAAGGCAGCTCACTGGACTGCAACTGACACTGATGCTCGAAAGTGTGGGTATCAAAC
1>111111B>BF1AEEGGGGGHGG0EEHHHHHHGGFGHHHHGG/EEEGGGGBGG/1BF/GGDFHFGHHHHHGFFHBGHEBBEE>EEGFFH?ECEGBFHHFB1BBBGGGHHFHBDGHFHGGGFHH1GHHDG2DHHHGGEGGGGFHGFGGGHGHHHHGGGGGHHHHHHHHHHCHHHG09:AGGGGBFGGGGGGGGGGGFFFFFFFFFB/-BFFEBBBFFFFFFEFFFBFFFBF?FFF/;BFBFFBFFFFF#
品質情報は、各塩基が PCR で生じたエラー率 $P$ から計算された Q 値を指す。Q 値は以下のように計算される。
$$
Q =-10 \times log_{10}P
$$
FASTQ 形式では、Q 値に対応する1つの文字が、各塩基に割り当てられて記録されている。
Qiime2 の成果物(artifact)
Qiime2 には、生データからインポートされた中間成果物(qzaファイル)と、それをブラウザに表示できるように変換した可視化成果物(qzvファイル)がある。

本稿の解析過程で作られる qza ファイルの中で、重要なものを以下にまとめた。ただし、PICRUSt2 analysis による成果物は除いている。

各ファイルの説明
-
demux.qza
各サンプルのFASTQ データをまとめたファイル。 -
rep-seqs.qza
データセット中に存在する read の塩基配列をまとめたファイル。識別用の ID も割り当てられている。 -
table.qza
データセット中に存在する塩基配列の read 数をまとめたファイル。各塩基配列は ID によって区別されており、塩基配列の中身は記録されていない。 -
silva-132-99-nb-classifier.qza
16S rDNA の塩基配列をまとめたデータベース。 -
taxonomy.qza
rep-seq.qza の塩基配列が、データベースにおけるどの細菌に属するのかをまとめたファイル。
Qiime2 のインストール
環境
macOS Catalina 10.15.1
conda にインストール
既に anaconda もしくは miniconda をインストールしている場合、Qiime2 (2019.7.ver.)の仮想環境を以下のコマンドで conda に構築することができる。公式サイトはこちら。
wget https://data.qiime2.org/distro/core/qiime2-2019.7-py36-osx-conda.yml
conda env create -n qiime2-2019.7 --file qiime2-2019.7-py36-osx-conda.yml
rm qiime2-2019.7-py36-osx-conda.yml
conda の仮想環境を activate する。
source activate qiime2-2019.7
qzvファイルは次のコマンドでブラウザに表示できる。
qiime tools view ファイル名.qzv
docker にインストール
miniconda や anaconda をインストールしていない場合は、docker で Qiime2 の仮想環境を構築することもできる。まず、docker image をダウンロードする。
docker pull qiime2/core:2019.7
次のコマンドで、docker image からコンテナを作成し、Qiime2 を起動する。
docker run -t -i -v $(pwd):/data qiime2/core:2019.7 /bin/bash
ただし、docker では qiime tools view ファイル名.qzv というコマンドを使用できず、ローカルで qzv ファイルをブラウザに表示させる手段がない。そこで、ローカルホストに可視化用のサーバー(Qiime2 viewer)を構築する。
まず、GitHub からQiime2 viewerをダウンロードする。
git clone https://github.com/qiime2/q2view.git
次に、ファイルのルートディレクトリで次のコマンドを実行すると、qiime2 viewer が build される。
npm install
npm run build
そして、次のコマンドで qiime2 viewer を起動する。
npm start
この状態で http://localhost:4242/ にアクセスすると、ブラウザに viewer が表示される。
生データのインポート
次のように、ダウンロードした生データを1つのフォルダにまとめておこう。
今回は、hmp2 というフォルダに生データをまとめたので、hmp2 フォルダで以下のコマンドを実行すると、Manifest.csv が得られる。
ls */* | \
awk -F'.' -v P="$(pwd)" 'BEGIN { print "sample-id,absolute-filepath,direction" } \
{if(NR%2==0){print $3","P"/"$0",reverse"}else{print $3","P"/"$0",forward"}}' > ManifestFile.csv
sample-id,absolute-filepath,direction
206534,/Users/home/hmp2/206534/HMP.2.206534.1.fq,forward
206534,/Users/home/hmp2/206534/HMP.2.206534.2.fq,reverse
206536,/Users/home/hmp2/206536/HMP.2.206536.1.fq,forward
206536,/Users/home/hmp2/206536/HMP.2.206536.2.fq,reverse
206538,/Users/home/hmp2/206538/HMP.2.206538.1.fq,forward
206538,/Users/home/hmp2/206538/HMP.2.206538.2.fq,reverse
(後略)
awkコマンドは、取得したレコードの各行に対して指定の処理を実行できる。
詳細を以下にまとめた。
| オプションなど | 機能 |
|---|---|
| -F | レコードの区切り記号を指定 |
| Begin | awk コマンドによる処理を実行する前の命令を指定 |
| if(NR%2==0) | 行数が 2 で割り切れる行でのみ実行することを指定 |
| else | if( ) が成立しない行でのみ実行することを指定 |
| Manifest.csv に出力するテキストを指定 | |
| $0 | テキストファイル全体を表示 |
| $3 | 取得したテキストファイルの 3 区画目を表示 |
ManifestFile.csv をもとにインポートする。
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path ManifestFile.csv \
--output-path demux.qza \
--input-format PairedEndFastqManifestPhred33
各オプションに代入する値を以下にまとめた。PHRED は、エンコードの方式を指す。FASTQ データに記載されている品質情報が、Q 値に 33 を加えた値であれば 33、64 を加えた値であれば 64 を選択する。両方を試してみて、解析エラーが表示されないモードを利用するとよい。
| Read type | PHRED | --type に代入 | --input-format に代入 |
|---|---|---|---|
| Single Ends | 33 | 'SampleData[SequencesWithQuality]' | SingleEndFastqManifestPhred33 |
| Single Ends | 33 | 'SampleData[SequencesWithQuality]' | SingleEndFastqManifestPhred64 |
| Pair Ends | 64 | 'SampleData[PairedEndSequencesWithQuality]' | PairedEndFastqManifestPhred33 |
| Pair Ends | 64 | 'SampleData[PairedEndSequencesWithQuality]' | PairedEndFastqManifestPhred64 |
公式サイトでは、--input-format にPairedEndFastqManifestPhred33V2を代入しているが、Manifest.csv の形式が異なるだけである。
生データの denoising
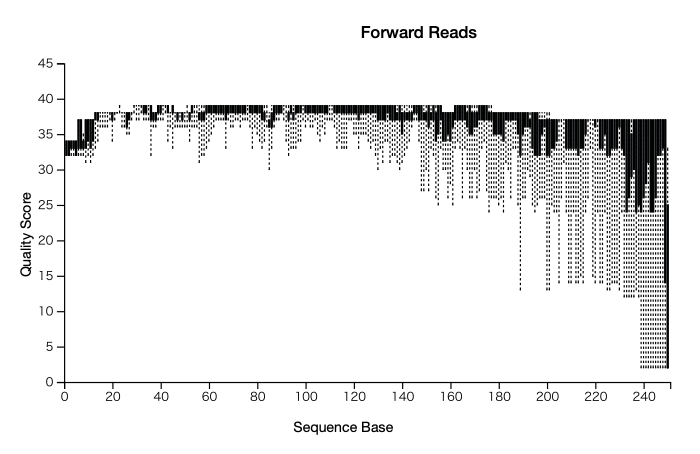
生データには、PCR 増幅時に生じたエラー配列が含まれている。まずは生データのクオリティスコア(Q値)を可視化して、配列の精度を確認してみる。
qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
ペアエンドシーケンスの場合、forward reads と reverse reads の Q 値がそれぞれ表示される。一般に Q 値は 3'末端から5'末端にかけて低下し、さらに reverse reads の方が低い。PCR 触媒は forward reads の 3'末端から順番に消耗していくため、徐々に精度は低下する。
アンプリコンには、真の配列以外に PCR で生じるエラーが含まれている。以下の概念図では、各配列の相同性と存在量を円の位置と大きさで表している。
OTU
16S rDNA のアンプリコン配列が一定の閾値以上で一致する集団を OTU という。PCR のエラー配列と真の配列を区別できないことがある。
そこで、PCRのエラー配列を修正あるいは除去し、真の配列から得られる細菌の分類単位を sub-OTU といい、従来の OTU と区別する。sub-OTU は DADA2 もしくは Deblur というパッケージで作成できる。特に DADA2 で得られる sub-OTU は、ASV (amplicon sequence varient) と呼ばれる。
DADA2
DADA2 では、エラー配列を真の配列に修正し、加算している。どのように修正するかをエラーモデルの学習によって推定している。

qiime dada2 denoise-paired \
--i-demultiplexed-seqs demux.qza \
--p-trunc-len-f 230 \
--p-trunc-len-r 220 \
--p-trim-left-f 0 \
--p-trim-left-r 0 \
--p-trunc-q 2 \
--p-n-reads-learn 1000000 \
--p-max-ee-f 2.0 \
--p-max-ee-r 2.0 \
--p-n-threads 4 \
--o-table table.qza \
--o-denoising-stats stats.qza \
--o-representative-sequences rep-seqs.qza \
--verbose
-
--p-trunc-len-f--p-trunc-len-r
マージに使用する read の長さを決める。demux.qzv を確認し、Q 値が低下する後半の配列を除去したい場合は短く設定すると良い。ただし forward reads と reverse reads がマージするには最低でも 20bp 以上のオーバーラップ領域が必要とされている。例えば 255bp の V4 領域は合計 275 bp なければマージされない。V3 領域も含めた生データには、最低でも合計 500 bp 程度が必要である。
-
--p-trim-left-f--p-trim-left-r
3'末端側で除去する read 長を指定する。アダプター配列などを除去する場合に用いる。
--p-trunc-q
Read の始まりから 1 塩基ずつ Q 値を調べ、この値より Q 値が高い最初の塩基を見つける。その塩基より先の配列を全てトリミングする。したがって、クオリティスコアの低い read の場合、この値が高いと塩基長が足りずにマージしなくなることがある。
この値より Q 値が低い塩基は --p-trunc-len-f や --p-trunc-len-r で設定した塩基長に到達するより前に見つかることもある。その場合 --p-trunc-len-f や --p-trunc-len-r で設定した塩基より短くトリミングされる。
-
--p-max-ee-f--p-max-ee-r
Read の始まりから 1 塩基ずつ P 値(Q 値ではない)を加算し、この値を超えた塩基より先の配列をトリミングする。--p-trunc-q と対照的に、クオリティスコアの低い read の場合、この値が低いと塩基長が足りずにマージしなくなることがある。
品質の低い塩基が続くと --p-trunc-len-f や --p-trunc-len-r で設定した塩基長に到達するより前に見つかることもある。その場合 --p-trunc-len-f や --p-trunc-len-r で設定した塩基より短くトリミングされる。
--p-n-reads-learn
エラーモデルの学習に要する read 数を指定する。小さいと処理が早く終わるがエラーモデルの精度が落ちる。
--p-n-threads
スレッド数を設定する。高いと処理が早く終わるが、それまで他の動作が鈍くなる。
--o-table
各配列の存在量データを出力する。塩基の並びは含まれない。
--o-representative-sequences
各配列の塩基データを出力する。
--o-denoising-stats
DADA2 の各工程で入力された read 数を出力する。

| 項目 | 内容 |
|---|---|
| Input | 入力された read 数 |
| Filtered | トリミング後の read 数。Q 値の基準が厳しいほど減少する。 |
| Denoised | Denoising 後の read 数。エラーモデルに不寛容であるほど減少する。 |
| Merged | マージ後の read 数。オーバーラップ領域が 20 bp 以下だと極端に減少する。 |
| Non-chimeric | 最終生成 read 数。PCR の精度が低いと減少する。 |
Deblur
Deblur にはペアエンドシーケンスのマージが実装されていないため、事前に Vsearch で merge させておく。
トリミング長は自動で設定される。
qiime vsearch join-pairs \
--i-demultiplexed-seqs demux.qza --o-joined-sequences demux-joined.qza
Deblur では、エラー配列を修正せずに除去する。DADA2 より配列の精度は高いが得られる read 数は少ない。
qiime deblur denoise-16S \
--i-demultiplexed-seqs demux-joined.qza \
--p-trim-length 250 \
--p-mean-error 0.005 \
--p-indel-prob 0.01 \
--p-indel-max 3.0 \
--p-jobs-to-start 36 \
--o-table table.qza \
--o-stats stats.qza\
--o-representative-sequences rep-seqs.qza \
--verbose
--p-trim-length
read 長を設定する。DADA2 と異なり、ここで指定した read 長しか出力されない。
--p-mean-error
1塩基あたりの平均塩基数を設定する。
--p-indel-prob
1塩基あたりの最大インデル(indel)数を設定する。エラー配列の挿入(insertion)と欠失(deletion)を略してインデルという。
-
--o-table--o-representative-sequences--o-denoising-stats
出力ファイルはいずれも DADA2 と共通である。
中間成果物の確認
DADA2 または Deblur を通して、微生物の存在量データ(カウントデータ)はtable.qzaに出力され、塩基配列データはrep-seqs.qzaに出力される。
両ファイルのデータは、sampleidによって紐づけられている。
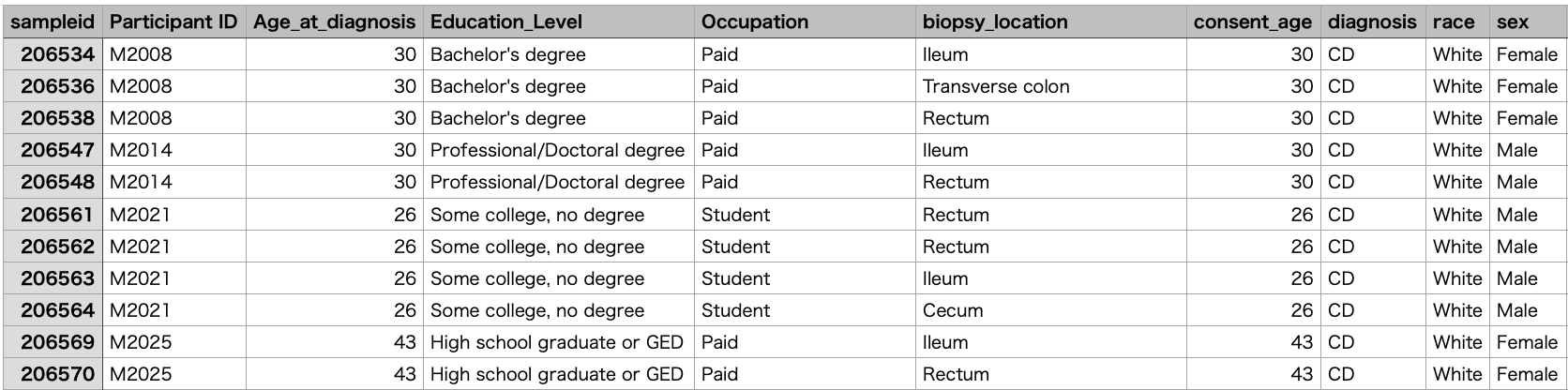
メタデータの作成
次のように、メタデータファイルを tsv 形式で作成する。Excel などの表計算ソフトで作成したデータシートを tsv 形式にエクスポートすると簡単に作れる。
sampleidは一番左のカラムに用意すること。詳しくはこちら。
sampleid Participant ID Age at diagnosis Education Level Occupation biopsy_location consent_age diagnosis race sex read
206534 M2008 30 Bachelor's degree Paid Ileum 30 CD White Female 33709.0
206536 M2008 30 Bachelor's degree Paid Transverse colon 30 CD White Female 28684.0
206538 M2008 30 Bachelor's degree Paid Rectum 30 CD White Female 15375.0
(後略)
サンプルの抽出
多変量解析には、read 数が足りず環境中の菌叢を正しく反映しているとはいえないサンプルは適さないため、除外する必要がある。
例えば、read 数 1500 以上のサンプルの抽出は、次のコマンドで実施できる。
qiime feature-table filter-samples \
--i-table table.qza \
--p-min-frequency 1500 \
--o-filtered-table selected_table.qza
さらにselected_table.qzaは、Metadata.tsv における特定の条件を満たすサンプルのみ抽出することができる。抽出されたtable.qzaも、全体データから得られた塩基データrep-seqs.qzaや細菌分類データtaxonomy.qza(後述)と対応している。
qiime feature-table filter-samples \
--i-table selected_table.qza \
--m-metadata-file Metadata.tsv \
--p-where "[sex]='Male’” \
--o-filtered-table Male/table.qza \
--output-dir Male
qiime feature-table filter-samples \
--i-table selected_table.qza \
--m-metadata-file Metadata.tsv \
--p-where " [Age at diagnosis]<30 AND [sex]='Male'” \
--o-filtered-table DisagnosisU30Male/table.qza \
--output-dir DisagnosisU30Male
Taxonomy analysis
公式サイトから、以下のうちいずれかのデータベースをダウンロードする。
- Silva 132 99% OTUs full-length sequences
- Silva 132 99% OTUs from 515F/806R region of sequences
- Greengenes 13_8 99% OTUs full-length sequences
- Greengenes 13_8 99% OTUs from 515F/806R region of sequences
Silva は現在も更新し続けているデータベースである。生データが V3 または V4 領域の場合は 515F/806R region of sequences を使用し、他の領域の配列であれば full-length sequences を使用する。
Greengenes は現在更新されていないデータベースであり、新種の細菌は登録されていない。OTU で細菌を分類していた頃に使われていた。
特別な理由がなければ Silva をダウンロードするとよい。
塩基データrep-seqs.qzaから細菌分類データtaxonomy.qzaを作成する。
qiime feature-classifier classify-sklearn \
--i-classifier silva-132-99-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
存在量データtable.qzaと合わせて棒グラフを得る。
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file Metadata.tsv \
--o-visualization taxa-bar-plots.qzv
taxa-bar-plots.qzv の上部で棒グラフの表示を設定できる。各内容について以下で説明する。
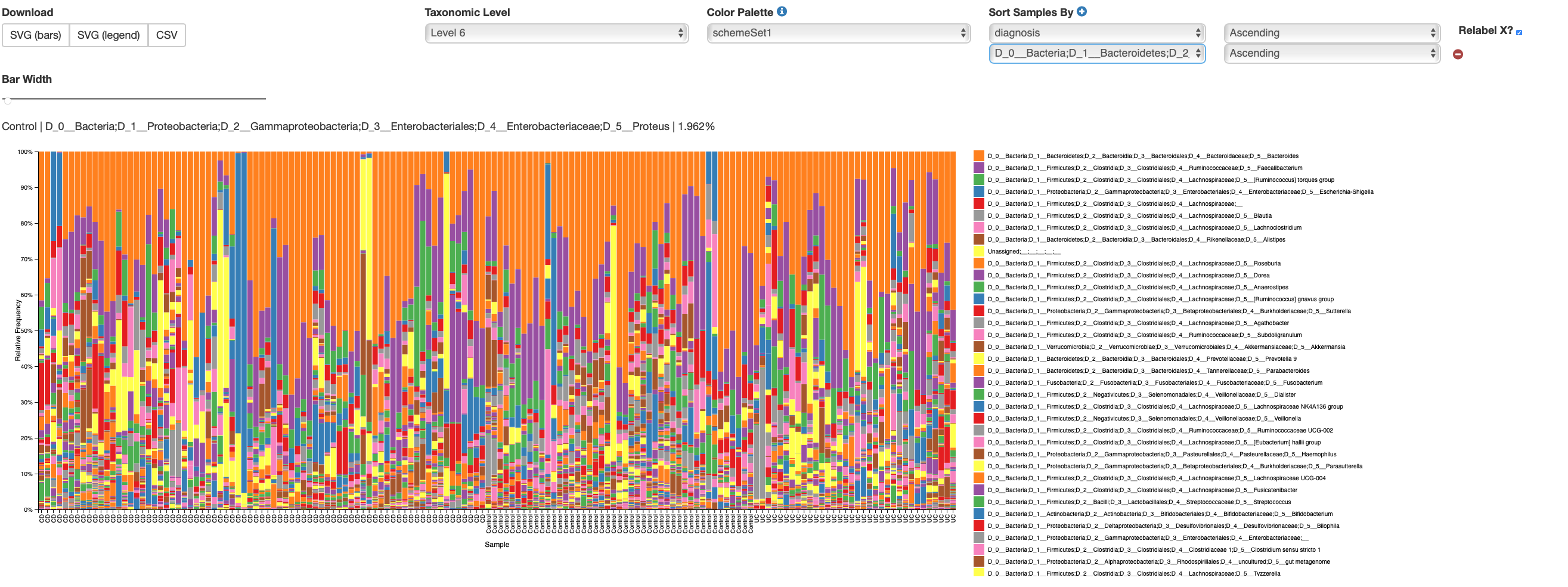
- Taxonomic Level
以下の表に対応している。なお、塩基データrep-seqs.qzaの分類は sub-OTU に基づくものであり、Species より細かい。
| Level1 | Level2 | Level3 | Level4 | Level5 | Level6 | Level7 |
|---|---|---|---|---|---|---|
| Kingdom:界 | Phylum:門 | Class:綱 | Order:目 | Family:科 | Genus:属 | Species :種 |
- Color Palette
棒グラフのカラーグラデーションを決める。
- Sort Samples By
棒グラフ整列時の優先順を決める。上記の例では sex(性別)、biopsy_location(生検部位)、diagnosis(診断名) の順に優先されて整列される。
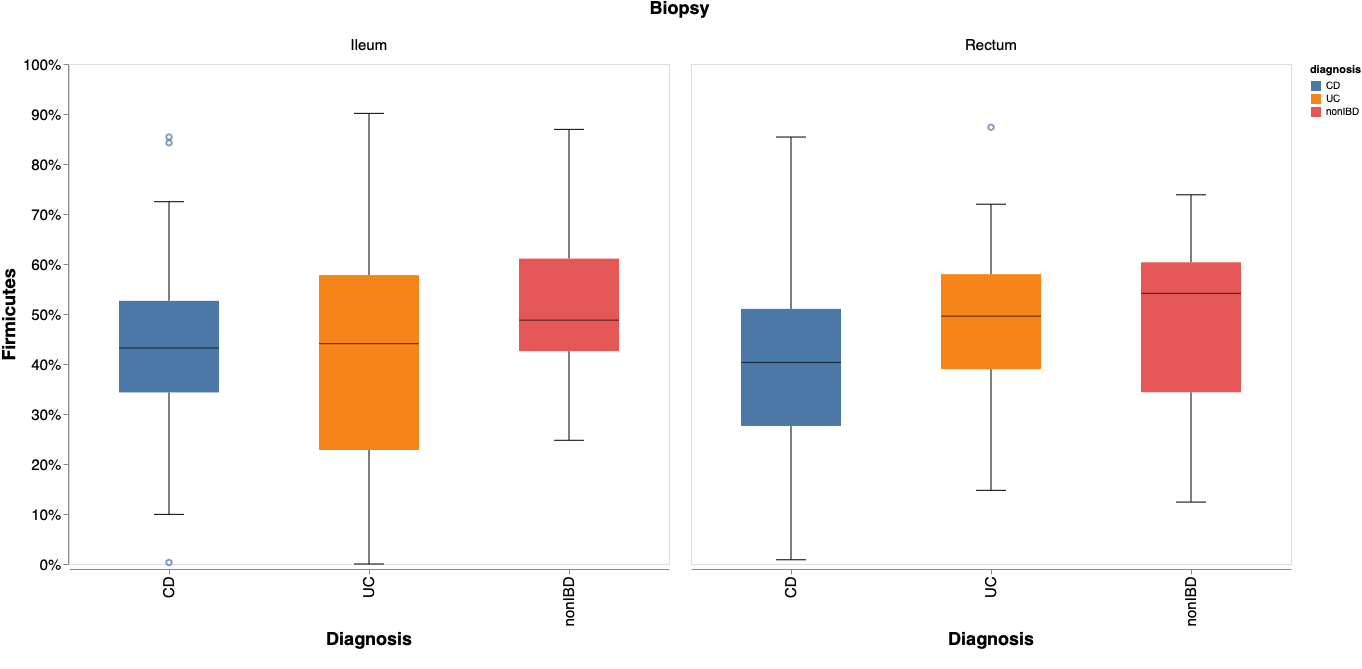
特定の細菌の構成率の分布を可視化する方法
以下のように、特定の細菌(例では Firmicutes 門)に着目して構成率の分布を表す方法は、こちらに紹介している。
PICRUSt2 analysis
菌叢代謝予測ツール PICRUSt2 は、次のコマンドでプラグインできる。
conda install q2-picrust2=2019.7 -c conda-forge -c bioconda -c gavinmdouglas
PICRUSt2 では、以下のデータベースに基づいて酵素群または代謝経路の存在量を予測する。
- EC (Enzyme Commision):酵素群
- KO (KEGG Orthology):酵素群
- MetaCyc Pathway :代謝経路(それぞれ数個〜数十個の酵素群を含む区分)
以下のコマンドで実行する。
qiime picrust2 full-pipeline \
--i-table table.qza \
--i-seq rep-seqs.qza \
--output-dir q2-picrust2_output \
--p-threads 4 \
--p-hsp-method pic \
--p-max-nsti 2 \
--verbose
EC や KO は区分が細かいため、対象の酵素がピンポイントで定まっている場合に利用するとよい。一方で MetaCyc Pathway は、関連する酵素群をまとめて分類しているため、系全体の現象を解釈するときに便利である。
データのエクスポート
table.qza を以下のコマンドで tsv 形式に変換する。
qiime tools export \
--input-path table.qza \
--output-path export
biom convert \
-i export/feature-table.biom \
-o export/feature-table.tsv \
--to-tsv
同様に、q2-picrust2_output/pathway_abundance.qzaを tsv 形式に変換する。
qiime tools export \
--input-path q2-picrust2_output/pathway_abundance.qza \
--output-path pathway_export
biom convert \
-i pathway_export/feature-table.biom \
-o pathway_export/feature-table.tsv \
--to-tsv
これにより、 qiime2 外部の R パッケージであるALDEx2などで多変量解析ができる。
Differential analysis
アンプリコン解析で得られるデータセットは、試料から抽出されたサンプルの read 数であって、試料に含まれる細菌の絶対的な存在量ではない。また、シーケンサーの性能や PCR の精度によって、サンプルの合計 read 数は変わる。したがって read 数はサンプル全体に対して何 % 占めるかという組成データに変換して比較されるべきである。組成データに対する多変量解析のツールとしてANCOM、Balance、ALDEx2などがある。
ANCOM
ANCOM (Analysis of composition of microbiomes) は、Qiime2 の公式チュートリアルでも紹介されている、組成データに対する多変量解析手法の 1 つである。
ANCOM モデル
あるデータセット全体に、$m$ 種の特徴量が存在し、群 $X$ に属するサンプル $i$ のカウントデータを $$X_i=(X_{i,1} \cdots \cdots, X_{i,m})$$ とおく。ここで、ある特徴量 $k$ に着目する。そして、$k$ を除く$m-1$ 種の特徴量 $l$ に対して、次の ANOVA モデルを総当たりで当てはめていく。
ln \frac{X_{i,k}+1}{X_{i,l}+1} =a_{k,l}+b_{k,l,X}+e_{k,l}
$a_{k,l}$ :特徴量 $k$ 、 $l$ における全サンプルの平均
$b_{k,l,X}$ :群 $X$ の効果を表す偏回帰係数
$e_{k,l}$ :過分散
モデルには、$$X_{i,l}=0$$ の場合でも計算できるように、pseudocount ($+1$) が組み込まれている。
同様に、群 $Y$ に属するサンプル $j$ のカウントデータを
$$Y_j=(Y_{j,1} \cdots \cdots, Y_{j,m})$$
においても、特徴量 $k$ に対して $m-1$ 通りの ANOVA モデルを当てはめていく。
ln \frac{Y_{j,k}+1}{Y_{j,l}+1} =a_{k,l}+b_{k,l,Y}+e_{k,l}
ANCOM では、
b_{k,l,X}=b_{k,l,Y}= 0
という帰無仮説について ANOVA を実施する。すなわち各特徴量は、自身以外の m-1 通りの特徴量を相手に検定される。群X、Yの間に定量的な差がある特徴量は、どんな特徴量を相手にしても帰無仮説が棄却されるため、最大で m-1 個の帰無仮説が棄却される。
ANCOM では、ある特徴量 k に対して棄却された帰無仮説の数を $W_k$ とする。この値が大きいほど、群間に定量的な差がある特徴量と判断される。
CLR:Centered log-ratio transformation について
ANCOM は定量的な差の有無を判定できるが、差の大きさは判定できない。そこで Qiime2 の ANCOM では、CLR という対数変換を用いて、差の大きさを独自で評価している。なお、ANCOM 専用の python パッケージに CLR は実装されていない。
さて m 個の特徴量に対する組成データの標本空間 $S^{m-1} $ は、
$$
S^{m-1}=\left\{ (x_{1}, x_{2}, \cdots,x_{m} ) \ | \ x_{i} \geq 0 \ (i=1,2, \cdots,m) \sum_{i=1}^{m}x_{i}= 1 \right\}
$$
である。位相幾何学では、この m-1 次元空間を単体空間(Simplex space) という。サンプル i のカウントデータ $$X_i=(X_{i,1} \cdots \cdots, X_{i,m})$$は、
$$
\frac{X_i}{\sum_{k=1}^{m}X_{i,k}}=(x_{1}, x_{2}, \cdots,x_{m})
$$
によって、単体空間 $S^{m-1} $ 上に変換される。この空間において、特徴量1と2の差は、
$$
x_{2}-x_{1}= \frac{X_{2}-X_{1}}{\sum_{i=1}^{m} X_{i}}
$$
となり、無関係の特徴量にも影響される。このような性質は、差の大きさを評価するのに適さないといえる。この問題は、$S^{m-1}$ を $m-1$ 次元のユークリッド空間 $R^{m-1} $ に変換(全単射)する、**有心対数比変換(Centered log-ratio transformation:CLR)**によって解決することが知られている。
組成データ $ x =(x_{1}, x_{2}, \cdots,x_{m} )$ に対する CLR とは
$$
clr(x)=\left\{log_e \frac{x_{1}}{g(X)}, log_e \frac{x_{2}}{g(X)}, \cdots,log_e \frac{x_{m}}{g(X)} \right\}
$$
である。ただし、$g(X)$ を幾何平均を
$$
g(x)=\sqrt[n]{x_{1}x_{2}\cdots x_{m}}
$$
とする。このとき、特徴量1と2の差は
$$
log_e \frac{x_{1}}{g(x)}-log_e \frac{x_{2}}{g(x)} = log_e \frac{x_{1}}{x_{2}}
$$
となり、幾何平均 $g(x)$ に依存しない。このような性質から、CLR は組成データを比較する有効な手段と考えられている。
コマンド
Qiime2 の解析パイプラインに沿って ANCOM を実施してみる。
今回は次のコマンドで CD 群と対象群のサンプルを抽出し、両軍の菌叢を比較する。
qiime feature-table filter-samples \
--i-table selected_table.qza \
--m-metadata-file Metadata.tsv \
--p-where "[diagnosis]='Control' OR [diagnosis]='CD'" \
--o-filtered-table CD_table.qza
上述の Taxonomy analysis で taxonomy.qza を求めている場合、sub-OTU で集計されている table.qza を genus (属)や family (科)などの上位階層で集計し直すこともできる。
次のコマンドでは、sub-OTU のカウントデータを genus レベルに合算している。
qiime taxa collapse \
--i-table All/CD_table.qza \
--i-taxonomy taxonomy.qza \
--p-level 6 \
--o-collapsed-table CD_L6_table.qza
ANCOM モデルにおける pseudocount を実施する。すなわち全てののカウントデータに対して $+1$ される。
qiime composition add-pseudocount \
--i-table CD_L6_table.qza \
--o-composition-table CD_L6_comp_table.qza
次のコマンドで、ANCOM を実施する。
qiime composition ancom \
--i-table CD_L6_comp_table.qza \
--m-metadata-file Metadata.tsv \
--m-metadata-column diagnosis \
--o-visualization CD_L6_ancom.qzv
出力結果の解釈
ANCOM の結果は、Volcano plot という形式で出力される。縦軸は、上で説明したとおり、棄却された帰無仮説の個数を表す。横軸は、対象群における CLR 値の平均値から、CD 群におけるそれを差し引いた値である。群間の差が小さい場合、横軸は 0 に近づく。

ちなみに 3 群以上の比較も実施できる。その場合、横軸は、ANOVA における CLR 値の F 値をとる。群間に差が存在しない場合、F 値は 0 に近づく。非負の値をとることに注意しよう。
各特徴量の詳細な W は、tsv 形式で表示される。このとき、全サンプルの W に対する累積分布関数から閾値が設定され、W が閾値以上の特徴量には統計的有意差があるとみなされ True と表示される。

True と判定された特徴量に対しては、ブラウザ上にカウントデータの四分位数が表示される。Pseudocounts の影響で、最低値は必ず 1.0 となる。

この例では、該当の特徴量の中央値に着目すると、CD 群では 1.0 だが、 Control 群では二桁である。いずれの genus も、CD 群で低下傾向にあることがわかる。
運用上の注意点
ANCOM は、多くの特徴量で群間に差がない場合に、群間に差がある一部の特徴量を索敵するのに適している。もし多くの特徴量で群間に差があると、帰無仮説
b_{k,l,X}=b_{k,l,Y}= 0
が棄却される k、l の組み合わせが無くなり、$W_k$ に差が付かなくなるためである。公式チュートリアルにも、 「ANCOM は25% 未満の特徴量に差があるデータセットを想定している」と書かれている。
ALDEx2
Qiime2 外部の R パッケージで解析する。背景やコマンドはこちらで紹介している。
Diversity analysis
Diversity analysis では、サンプルの多様性を表す指標(α-多様性とβ-多様性)をもとに、群間を比較する。
系統樹の作成
多様性指標の中でも、α 多様性指標である Faith’s Phylogenetic Diversity (Faith PD) と、β 多様性指標である Unifrac 距離は、微生物の系統樹に基づいて計算される。
まずは次のコマンドで、塩基配列データrep-seqs.qzaから系統樹rooted-tree.qzaを作成しておく。上述以外の指標は、系統樹を作成せずとも計算できる。
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
多様性指数の計算
以下の多様性指数は、次のコマンドによって core-metrics-results にまとめて出力できる。
- α 多様性指数
- Shannon index
- 観測された OTU の数
- Faith’s Phylogenetic Diversity (Faith PD)
- Pielou’s Evenness (Pielou の均衡度指数)
- β 多様性指数
- Jaccard 距離
- Bray Curtis 距離
- Unweighted Unifrac 距離
- Weighted Unifrac 距離
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table selected_table.qza \
--p-sampling-depth 1500 \
--m-metadata-file metadata.tsv \
--output-dir core-metrics-results
サンプル深度について
多様性指標は、サンプルの全 read 数に影響されやすい。例えば、1000 read のサンプルと 10000 read のサンプルは正しく比較できないことがある。そこで、サンプルからのランダム抽出で read 数を希薄化 (Rarefaction) することがある。希薄時の抽出 read 数をサンプル深度といい、上記コマンドでは--p-sampling-depth で 1500 に設定している。read 数がサンプル深度以下のサンプルは解析から外される。サンプル深度を小さくすると希少種が抽出されないことがあるが、サンプル深度を大きくすると解析対象のサンプル数が減ってしまう。解析から、どのような主張を導きたいかによってサンプル深度は決定する必要がある。
α 希薄化 (alpha rarefaction)
適切なサンプル深度を決定するために、様々なサンプル深度で α 多様性指数を計算する操作を**α 希薄化(alpha rarefaction)**という。
次のコマンドでは、最大深度 10000 read を 21 段階に区切り、500 read ごとに希薄化し α 多様性指数を求めている。
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 10000 \
--p-steps 21 \
--m-metadata-file Metadata.tsv \
--o-visualization alpha-rarefaction.qzv
Shannon index は、こちらで説明したとおり主要な種に影響を受けやすいため、サンプル深度が小さい段階から閾値に近い。下図では、CD 群、Control 群、UC 群の腸内細菌叢における shannon index を表しているが、1500 read 以上でほぼ一定である。このことから、1500 read までの抽出で腸内細菌の主要な種は含まれていると考えられる。

一方、下図の observed OTUs (一度でも観測された OTU の数)は、サンプル深度と共に増え続けている。これは、サンプル深度を大きくするほど希少種が抽出されるためである。この傾向は特に菌叢多様性の高い土壌サンプルなどで見られる。多様な種を抽出することを優先してサンプル深度を大きくすると、解析対象のサンプル数が減ってしまうので注意すること。

その他について
なお、こちらで紹介しているように、多様性指数には他の種類もあるため、必要に応じて個別に計算してみよう。
- Chao1
qiime diversity alpha \
--i-table selected_table.qza \
--p-metric 'chao1' \
--o-alpha-diversity core-metrics-results/chao1_vector.qza
- Simpson
qiime diversity alpha \
--i-table selected_table.qza \
--p-metric 'simpson' \
--o-alpha-diversity core-metrics-results/simpson_vector.qza
- Simpson Index of Evenness
qiime diversity alpha \
--i-table selected_table.qza \
--p-metric 'simpson_e' \
--o-alpha-diversity core-metrics-results/simpson_e_vector.qza
α 多様性指数の比較
- shannon index の比較
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/shannon_vector.qza \
--m-metadata-file Metadata.tsv \
--o-visualization core-metrics-results/shannon_significance.qzv
Shannon index を縦軸に、CD 群、Control 群、 UC 群の箱ひげ図が表示される。

さらに、クラスカル=ウォリス検定の結果が表示される。今回は、CD 群、Control 群、UC 群の 3 群比較であり、pairwise テストは 3 回実施されている。したがって、多重比較による第一種の過誤を防止するために、BH法で偽陽性率(FDR)を調整した q-value を参考にするのが良い。

- Observed OTUs の検定
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/observed_otus_vector.qza \
--m-metadata-file Metadata.tsv \
--o-visualization core-metrics-results/observed_otus_significance.qzv
- Faith’s Phylogenetic Diversity (Faith PD) の検定
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file Metadata.tsv \
--o-visualization core-metrics-results/faith_pd_significance.qzv
- Pielou’s Evenness (Pielou の均衡度指数) の検定
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/evenness_vector.qza \
--m-metadata-file Metadata.tsv \
--o-visualization core-metrics-results/evenness_significance.qzv
β 多様性指数の比較
β 多様性の有意差検定には、ノンパラメトリック分散分析として知られる PERMANOVA(PERmutational Multivariate ANalysis Of VAriance) を採用する。
PERMANOVA について
PERMANOVAでは、通常の分散分析と同様に、平均に対する二乗和(Sum of Squares:SS) から F 統計量を求める。
$$
F= \frac{ \frac{SS_T-SS_w}{a-1}}{\frac{SS_w}{N-a}} = \frac{ \frac{SS_a}{a-1}}{\frac{SS_w}{N-a}}
$$
$SS_T$ :全てのサンプル間の β 多様性指標の平均に対する二乗和
$SS_W$ :同一群内のサンプル間の β 多様性指標の平均に対する二乗和
$SS_A$ :異なる群に属するサンプル間の β 多様性指標の平均に対する二乗和
$N$ :サンプルの数
$a$ :群の数
群間の差が小さい場合、すなわち群間で菌叢に違いがない場合、$SS_A$ が小さくなり、F も小さくなる。一方で、菌叢の差が大きい群間ほど F は大きくなる。
ただし通常の F 検定とは異なり、F 分布を帰無分布と仮定していない。帰無分布には、サンプルをランダムに並べ替え (permutation) て計算された $F^H$ の分布が採用されている。Qiime2 ではデフォルトで 999 回の並べ替えシミュレーションを実施しており、その際の $F^H$ の分布をもとに検定している。
コマンド
検定コマンドでは、メタデータのどの項目で菌叢を比較するかを設定する --m-metadata-column が必須である。
- Jaccard 距離の比較
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/jaccard_distance_matrix.qza \
--m-metadata-file Metadata.tsv \
--m-metadata-column diagnosis \
--o-visualization core-metrics-results/jaccard_distance_significance.qzv \
--p-pairwise
- Bray Curtis 距離の比較
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/bray_curtis_distance_matrix.qza \
--m-metadata-file Metadata.tsv \
--m-metadata-column diagnosis \
--o-visualization core-metrics-results/bray_curtis_distance_significance.qzv \
--p-pairwise
- Unweighted Unifrac 距離の比較
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file Metadata.tsv \
--m-metadata-column diagnosis \
--o-visualization core-metrics-results/unweighted_unifrac_significance.qzv \
--p-pairwise
- Weighted Unifrac 距離の比較
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/weighted_unifrac_distance_matrix.qza \
--m-metadata-file Metadata.tsv \
--m-metadata-column diagnosis \
--o-visualization core-metrics-results/weighted_unifrac_significance.qzv \
--p-pairwise
β 多様性指標の成果物は下図のとおり。

Group significance plots では、「同一群内のサンプル間の β 多様性指標の箱ひげ図」が最も左に表示され、他は「異なる群に属するサンプル間の β 多様性指標の箱ひげ図」となる。
例では、CD 群(n=75)、Control 群(n=45)、UC 群(n=34) を比較している。CD 群に着目すると、「同一群内のサンプル間の β 多様性指標」は、75×74=2775 通り計算され、「Control 群に属するサンプル間の β 多様性指標」は、75×45=3375 通り計算され、「UC 群に属するサンプル間の β 多様性指標」は、75×34=2550 通り計算されている。
PERMANOVAでは、CD 群、Control 群、UC 群の 3 群を比較しており、pairwise テストは 3 回実施されている。したがって、多重比較による第一種の過誤を防止するために、BH法で偽陽性率(FDR)を調整した q-value を参考にするのが良い。