概要
ショットガンメタゲノム解析は、サンプル内に存在する細菌群集由来のゲノム全体を断片化し、それらをランダムにシーケンシングすることで群集全体の遺伝子組成を明らかにする手法である。16S rRNA 遺伝子などの特定の系統マーカー遺伝子をターゲットとするアンプリコン解析とは異なり、世界標準といえる手順は確立されていないが、いくつかの解析パイプラインは存在する。
本項では、ショットガンメタゲノムデータから細菌群集の組成や群集がもつ遺伝子を定量することができるパイプライン HUMAnN (Human microbiome project Unified Metabolic Analysis Network) のバージョン 3.0、すなわち HUMAnN3.0 によるショットガンメタゲノム解析について紹介する。HUMAnN では次世代シーケンスデータをもとに細菌群集がもつ酵素遺伝子を定量する。酵素の発現量ではなく、酵素を発現するポテンシャルを推定していることに注意しよう。
解析の流れ
本稿で紹介する解析手順は 3 段階で構成される。
1) BBtools によるペアエンドシーケンスのマージ
HUMAnN はシングルエンドシーケンスを対象にした解析手法であるため BBtools の BBmerge でペアエンドシーケンスをマージする。シングルエンドシーケンスデータを解析する場合、この工程はスキップしてよい。
2) KneadData によるホストゲノムのフィルタリング
コンタミしたホストゲノムをリファレンスデータベースに基づき除去する。
3) HUMAnN 3.0 による酵素遺伝子の定量
まず MetaPhlAn(Metagenomic Phylogenetic Analysis) で細菌群集組成を定量し、その情報をもとに細菌群集がもつ酵素遺伝子を定量する。
環境
- 機種: Mac mini
- メモリ: 64 GB
- OS: macOS Big Sur (バージョン 11.2.3)
※ 1TB のストレージが必要
テストデータのダウンロード
本稿では IBD multi'omics database に公開されている糞便のショットガンメタゲノムデータのうち 9 サンプル (21.7GB)をテストデータとして採用する。1サンプルあたり 2GB 以上のデータを扱うため、ファイル転送プロトコル (FTP) によるファイル転送アプリ Filezilla を用いて高速でダウンロードすることを推奨する。
ダウンロードページにあるように、以下の情報を入力して「クイック接続(Q)」を押すと Filezilla で公共データにアクセスできるようになる。
- ホスト(H): ftp.broadinstitute.org
- ユーザー名(U): public
- パスワード(W): hmp2_ftp
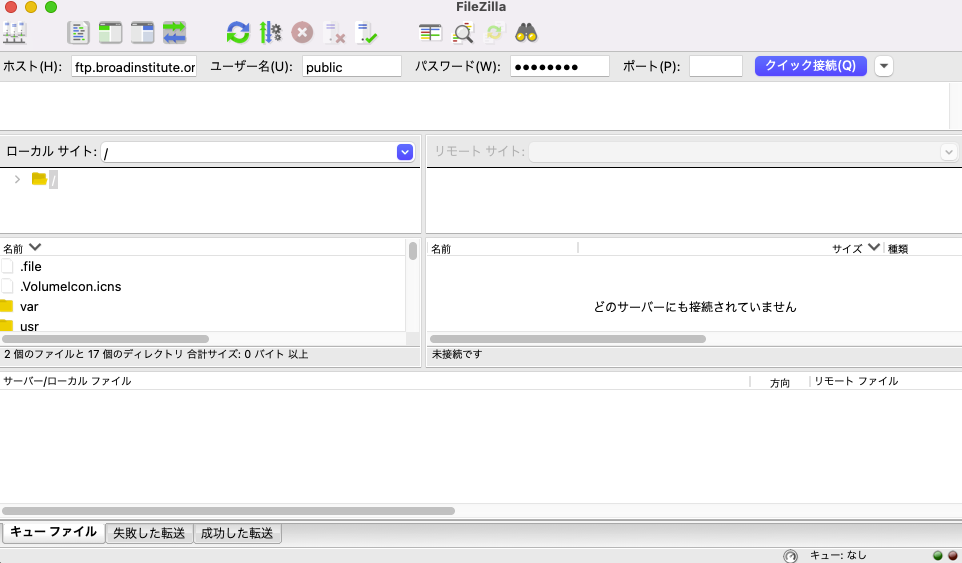
接続前の Filezilla は以下のとおり。ローカルサイトにはローカルサーバーのファイル構成が表示されている。アクセス前のためリモートサーバーは空である。
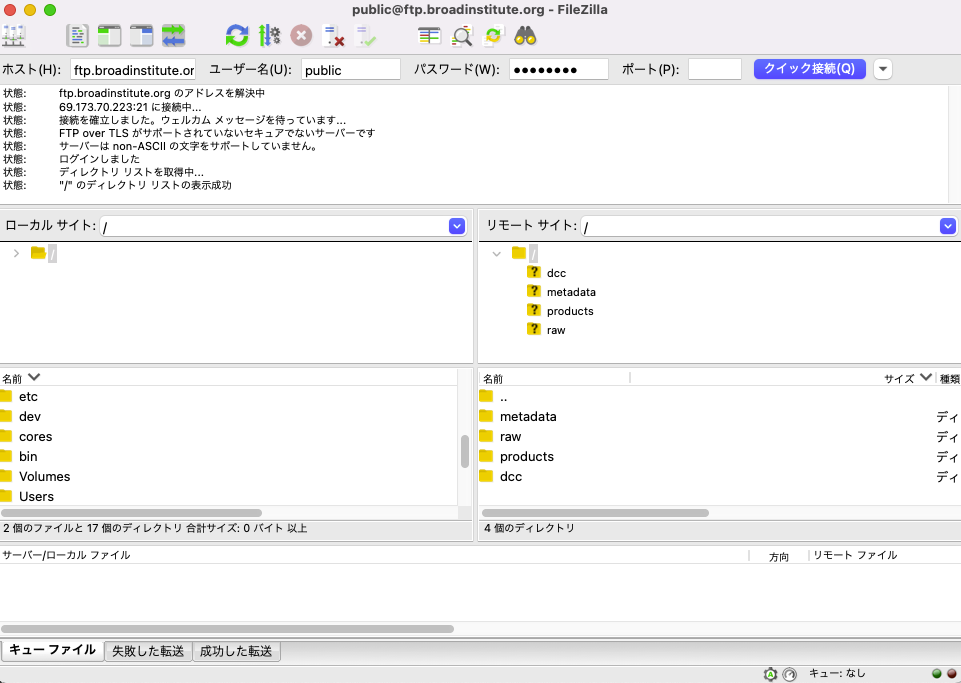
接続後の Filezilla は以下のとおり。IBD multi'omics database のフォルダ構成がリモートサイトに表示されている。
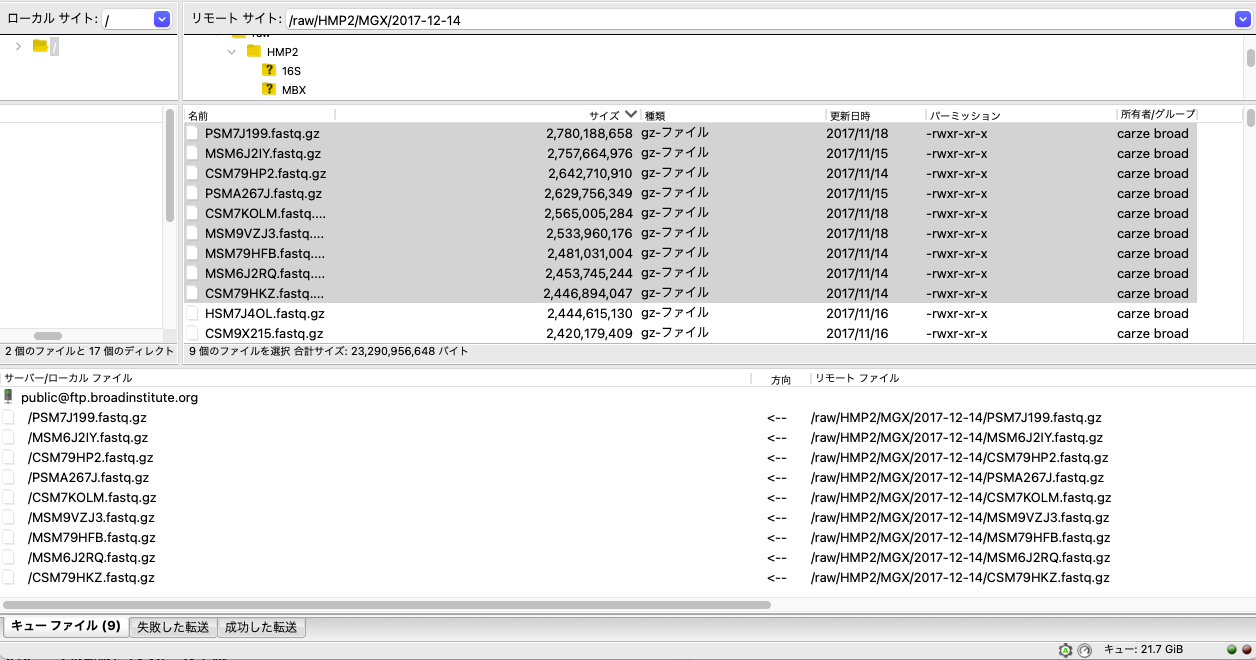
リモートサイトの /raw/HMP2/MGX/2017-12-14 にアクセスして、容量の大きい 9 ファイル(合計21.7 GB)をキューファイルにドラッグ & ドロップする。
ダウンロードデータの設置先をローカルサイトで設定し、キューファイルを右クリックして「キューを処理」を選択するとダウンロードが始まる。
BBtools によるペアエンドシーケンスのマージ
HUMAnN はシングルエンドシーケンスを対象にした解析手法であるため BBtools の BBmerge でペアエンドシーケンスをマージする。シングルエンドシーケンスデータを解析する場合、この工程はスキップしてよい。
インストール
Conda でインストールする。
conda install -c bioconda -y bbmap
ペアエンドシーケンスのマージ
BBtools の BBmerge を用いてペアエンドシーケンスをマージする。forward.fastq.gz と reverse.fastq.gz をマージするコマンドを以下に示す。
bbmerge.sh in1=forward.fastq.gz in2=reverse.fastq.gz out=output.fastq
Python を用いて複数のサンプルを自動解析するスクリプトを紹介する。ここでは *_R1.fastq.gz を forward sequence をとし、*_R2.fastq.gz を reverse sequence とする。以下に例 /data を示す。
Sample1_R1.fastq.gz
Sample1_R2.fastq.gz
Sample2_R1.fastq.gz
Sample2_R2.fastq.gz
Sample3_R1.fastq.gz
Sample3_R2.fastq.gz
Sample4_R1.fastq.gz
Sample4_R2.fastq.gz
以下のスクリプトで /data複数サンプルの自動実行が可能である。
import subprocess
import struct
# フォルダ名入力
folder = 'data'
# サンプル名取得
cmd = 'ls {0}/*.fastq.gz'.format(folder)
tmp = subprocess.check_output(cmd, shell=True, encoding='utf8').split('\n')[:-1]
sn = list(set(map(lambda w: w.split('/')[-1].split('_')[0],tmp)))
for s in sn:
# ファイル名取得
cmd = 'ls {0}/{1}*.fastq.gz'.format(folder,s)
fn = subprocess.check_output(cmd, shell=True, encoding='utf8').split('\n')[:-1]
# 実行
cmd = 'bbmerge.sh in1={0} in2={1} out=bbmerge/{2}/{3}.fastq'.format(fn[0],fn[1],folder,s)
subprocess.call(cmd,shell=True, encoding='utf8')
KneadData によるホストゲノムのフィルタリング
HUMAnN 開発グループ Huttenhower Lab の Github にも記載されているが、ショットガンメタゲノム解析ではサンプルに含まれているホストゲノムを事前にフィルタリングすることが推奨されている。本項では同グループが開発した KneadData を用いてホストゲノムを除去する。
解析の流れ
KneadData は以下の流れでフィルタリングする。まず Trimmomatic でクオリティの低い塩基やアダプター配列を除去する。次に Bowtie 2 でホストゲノムのリファレンスにマッピングした read を除去する。
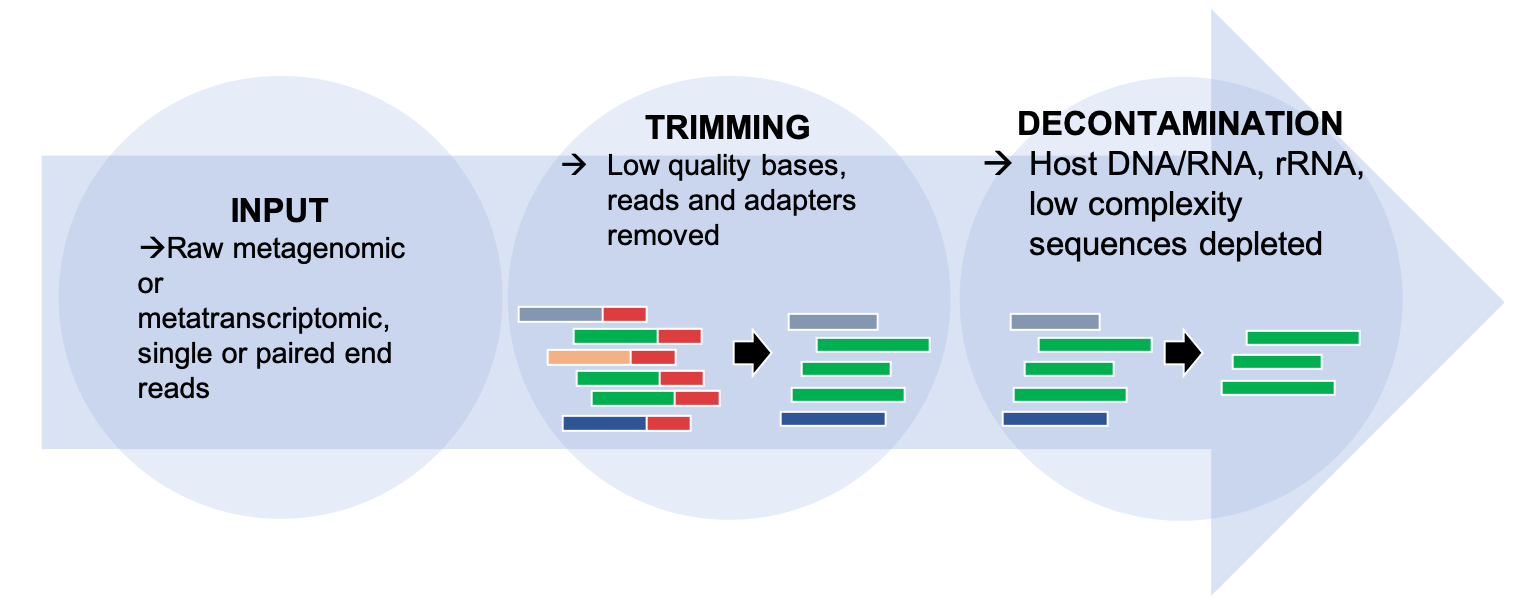
参照: 公式チュートリアル
上図において decontamination する前に TRF(Tandem Repeats Finder) を用いて反復配列を除去することもできる。この手順も本項では紹介する
インストール
pip で最新版をインストールする。
pip install kneaddata
TRF(Tandem Repeats Finder) のインストール
オプジョンで TRF(Tandem Repeats Finder) をインストールすることができる。KneadData の Github にある setup.py を実行するとよい
python setup.py install
Bowtie 2 のインストール
公式ドキュメントによれば、KneadData インストール時に Trimmomatic と Bowtie 2 は自動でインストールされるはずである。しかし bowtie2 と入力して以下のエラーが生じた場合、Bowtie 2 を再インストールする必要がある。
Error message returned from bowtie2 :
dyld: Library not loaded: @rpath/libtbb.dylib
Referenced from: /Users/ユーザー名/miniconda3/envs/humann/bin/bowtie2-align-s
Reason: image not found
brew でインストールする。bowtie2 の PATH が通っていれば KneadData で実行可能である。
brew install bowtie2
Docker でインストール
Docker で仮想環境を構築することもできる。最初から TRF もインストールされる。
最新版を随時確認して pull しよう。
docker pull biobakery/kneaddata:0.10.0
docker run --name kneaddata -itv $(pwd):/tmp biobakery/kneaddata:0.10.0
ホストゲノムのリファレンスをダウンロード
次に、コンタミしているホストゲノムのリファレンスをダウンロードする。ヒトゲノムの場合、公式ドキュメントによれば以下のコマンドでダウンロードできるようである。
kneaddata_database --download human_genome bowtie2 kneaddata_human_genome_bowtie2_reference_db/
しかし、ファイル容量が大きいせいか CRITICAL ERROR が表示されることも多い。
CRITICAL ERROR: Unable to download and extract from URL: http://huttenhower.sph.harvard.edu/kneadData_databases/Homo_sapiens_hg37_and_human_contamination_Bowtie2_v0.1.tar.gz
この場合、エラーメッセージの URL を wget で取得するとよい。ダウンロード後はファイルを展開しておくこと。
wget http://huttenhower.sph.harvard.edu/kneadData_databases/Homo_sapiens_hg37_and_human_contamination_Bowtie2_v0.1.tar.gz
他のゲノムのリファレンスも http://huttenhower.sph.harvard.edu/kneadData_databases/ にアクセスして選んでもよい
フィルタリングの実行
上記の工程が完了すれば、Trimmomatic、TRF、Bowtie 2 からなる一連の解析を以下のコマンドで実行できる。なおホストゲノムのリファレンスを wget で取得した場合、フォルダを展開しないと読み込まれない。TRF と Bowtie 2 は並列で実行されるため、スレット数を高く設定しておくと解析時間を短縮できる。ちなみに --bypass-trf をつけると TRF による反復配列の除去が省略される。
kneaddata --input INPUT.fastq.gz --reference-db Homo_sapiens_hg37_and_human_contamination_Bowtie2_v0.1 --output 出力先 -t スレッド数
正規表現を用いた全ファイル一括実行は以下のようにするとよい。テストデータ 9 サンプルの場合、12 スレッドで完了までに 4 時間かかる。*.trimmed.fastq と *.repeats.removed.fastq は中間成果物であるため、必要のない場合は削除するとよい。
for f in *.fastq.gz;do kneaddata -i $f -db Homo_sapiens_hg37_and_human_contamination_Bowtie2_v0.1 -o kneaddata_res/ -t スレッド数 ;\
rm kneaddata_res/*.trimmed.fastq ;\
rm kneaddata_res/*.repeats.removed.fastq ;done
成果物の説明
- INPUT_kneaddata.trimmed.fastq : Trimmomatic で低クオリティ read が除去された残りの read
- INPUT_kneaddata.repeats.removed.fastq : TRF で反復配列が除去された残りの read
- INPUT_kneaddata_hg37dec_v0.1_bowtie2_contam.fastq : Bowtie2 で除去されたホストゲノム read
- INPUT_kneaddata.fastq : 最終成果物
HUMAnN 3.0 によるメタゲノム解析
HUMAnN3.0 では、まず MetaPhlAn(Metagenomic Phylogenetic Analysis) を用いて細菌群集組成を定量し、その情報をもとに細菌群集がもつ酵素遺伝子を定量する。最終目的が細菌群集組成の定量であれば MetaPhlAn を実施すればよい。
解析の流れ
解析のワークフローは基本的に HUMAnN 2.0 と同じである。HUMAnN 3.0 公式チュートリアルにも HUMAnN 2.0 のワークフローが載っている。
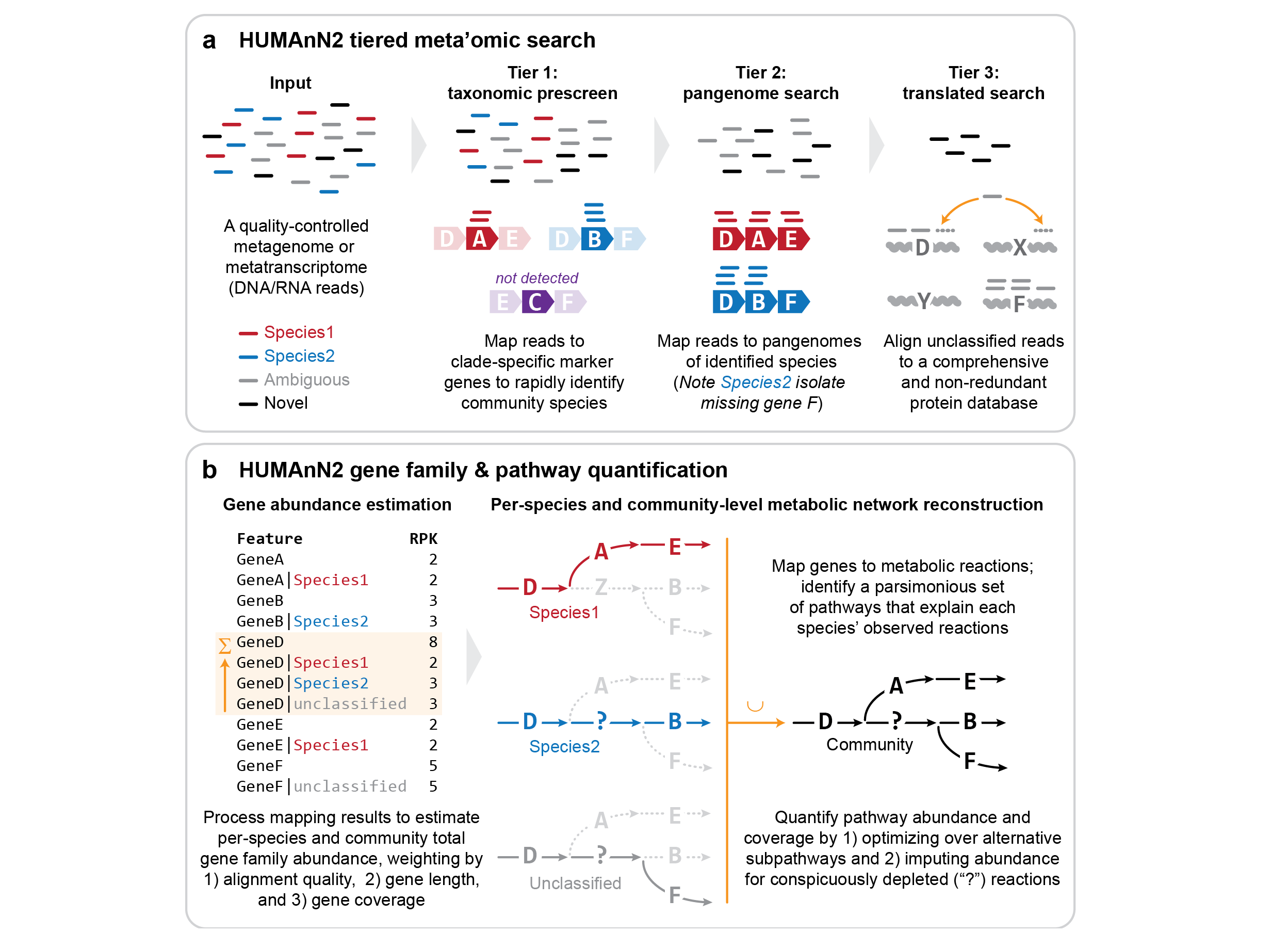
参照: 公式チュートリアル
a: HUMAnN2 tiered meta'omics search
HUMAnN 2.0 および 3.0 では、ショットガンメタゲノムデータを三段階でマッピングしている。
- Tier 1: まず MetaPhlAn を用いて、細菌および古細菌、ウイルス、真核生物のリファレンスゲノムから同定された独自のクレード特異的マーカー遺伝子に基づいて細菌群集の構成を明らかにする。
- Tier 2: 次に Bowtie 2 を用いて、MetaPhlAn で同定された細菌および古細菌、ウイルス、真核生物のゲノム(パンゲノム)に対してマッピングして、細菌群集がもつ酵素を同定する。
- Tier 3: 最後に Diamond を用いて、Bowtie 2 でマッピングされなかった配列に対してタンパク相同性解析を実施して、細菌群集がもつ酵素を同定する。
b: HUMAnN2 gene family & pathway quantification
マッピングされる酵素の種類は膨大であるため、近縁の酵素を遺伝子ファミリーや代謝経路としてまとめて集計する。
データベースと成果物について
HUMAnN 2.0 のものではあるが、解析のワークフローは以下の図でも紹介されている。
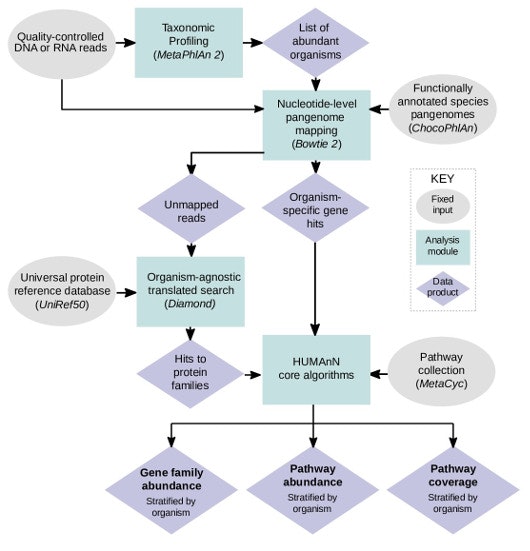
参照:
データベース
HUMAnN 2.0 および HUMAnN 3.0 では解析の過程で 3 つのデータベースを使用する。
- 酵素の同定に必要なパンゲノムデータベース ChocoPhlAn
- タンパクの相同性検索に必要なタンパクデータベース UniRef 90 (UniRef 50 を使用することもある)
- 代謝経路の集計に必要な代謝経路データベース MetaCyc
成果物
HUMAnN 2.0 および HUMAnN 3.0 では一連の解析で 3 つの成果物が得られる。
-
Gene family abundance: 遺伝子ファミリー(近縁の酵素をまとめたもの)の存在量 -
Pathway abundance: 代謝経路(近縁の遺伝子ファミリーをまとめたもの)の存在量 -
Pathway coverage: 代謝経路のカバレッジ(経路を構成する酵素すべてが存在する確率)
参照:
Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, et al. (2012) Metabolic Reconstruction for Metagenomic Data and Its Application to the Human Microbiome. PLoS Comput Biol 8(6): e1002358. https://doi.org/10.1371/journal.pcbi.1002358
このほか、中間ファイルの metaphlan_bugs_list には細菌群集構成が記録される(後述)。
コンパイラの alias 設定
HUMAnN 3.0 のインストールで C 言語をコンパイルする際、コンパイラ(GCC)の alias の設定が必要である。
brew で GCC のインストールおよびアップグレードをしておこう。
brew install gcc
brew upgrade gcc
ターミナル起動時に読み込まれる ~/.bash_profile に alias を追加する。
インストールした GCC のバージョンが 9.2.0_2 だった場合、以下のコマンドを実行する。
echo 'alias gcc="/usr/local/Cellar/gcc/9.2.0/bin/gcc-9"' >> ~/.bash_profile
HUMAnN 3.0 のインストール
pip で最新版をインストールする。
pip install humann==v3.0.0.alpha.4 --no-binary :all:
Docker でインストール
Docker で仮想環境を構築することもできる。
最新版を随時確認して pull しよう。
docker pull biobakery/humann:3.0.0.a.4.plus.utility.dbs
docker run --name humann3 -itv $(pwd):/tmp docker.io/biobakery/humann:3.0.0.a.4.plus.utility.dbs
リファレンスデータベースのダウンロード
HUMAnN 3.0 のワークフローでは、パンゲノム、タンパク、代謝経路という 3 つのデータベースが必要である。各データベースは個別でダウンロードする必要がある。
公式マニュアルによれば、以下のコマンドでダウンロードすることできる。
humann_databases --download chocophlan full $INSTALL_LOCATION ## パンゲノムデータベース
humann_databases --download uniref uniref90_diamond $INSTALL_LOCATION ## タンパクデータベース
humann_databases --download utility_mapping full $INSTALL_LOCATION/ ## 代謝経路データベース
しかし Difficulty downloading databases in humann3 という Forum にあるように、現在これらのコマンドは使用できない。そこで開発グループの dropbox から wget で入手する。経緯の詳細は Forum に記載されている。
wget https://www.dropbox.com/s/das8hdof0zyuyh8/full_chocophlan.v296_201901.tar.gz ## パンゲノムデータベース
wget https://www.dropbox.com/s/yeur7nm7ej7spga/uniref90_annotated_v201901.tar.gz ## タンパクデータベース
wget http://huttenhower.sph.harvard.edu/humann2_data/full_mapping_v201901.tar.gz ## 代謝経路データベース
(代謝経路データベースのみは wget を使わなくてもダウンロードできるようである。)
HUMAnN 2.0 との違い
HUMAnN 3.0 の主な更新点を以下に挙げる。MetaPhlAn のバージョンアップや、各種データベースのアップデートなどが行われたようだ。
- Designed in tandem with MetaPhlAn 3.0.
- Based on UniProt/UniRef 2019_01 sequences and annotations.
- Contains 2x more species pangenomes and 3x more gene families (vs. v2.0).
- Removed version number from executables (just metaphlan and humann now).
参照:
MetaPhlAn のバージョンアップによって以下の改善がなされた。ChocoPhlAn が更新されたり、ウイルスのプロファイリングが可能になったりしたようだ。
- New MetaPhlAn marker genes extracted with a newer version of ChocoPhlAn based on UniRef
- Estimation of metagenome composed by unknown microbes with parameter --unknown_estimation
- Automatic retrieval and installation of the latest MetaPhlAn database with parameter --index latest
- Virus profiling with --add_viruses
- Calculation of metagenome size for improved estimation of reads mapped to a given clade
- Inclusion of NCBI taxonomy ID in the ouput file
- CAMI (Taxonomic) Profiling Output Format included
- Removal of reads with low MAPQ values
参照:
Config の設定
HUMAnN 3.0 では、解析時のパラメータを humann_config で設定できる。まず humann_config の内容を確認してみよう。
$ humann_config
HUMAnN Configuration ( Section : Name = Value )
database_folders : nucleotide = パンゲノムデータベースのフォルダに設定したい
database_folders : protein = タンパクデータベースのフォルダに設定したい
database_folders : utility_mapping = 代謝経路データベースのフォルダに設定したい
run_modes : resume = False
run_modes : verbose = False
run_modes : bypass_prescreen = False
run_modes : bypass_nucleotide_index = False
run_modes : bypass_nucleotide_search = False
run_modes : bypass_translated_search = False
run_modes : threads = 1
alignment_settings : evalue_threshold = 1.0
alignment_settings : prescreen_threshold = 0.01
alignment_settings : translated_subject_coverage_threshold = 50.0
alignment_settings : translated_query_coverage_threshold = 90.0
alignment_settings : nucleotide_subject_coverage_threshold = 50.0
alignment_settings : nucleotide_query_coverage_threshold = 90.0
output_format : output_max_decimals = 10
output_format : remove_stratified_output = False
output_format : remove_column_description_output = False
データベースを wget でダウンロードした場合、config の内容は更新されないため、データベースフォルダの PATH を自分で入力する必要がある。
humann_config --update database_folders nucleotide パンゲノムデータベースの PATH
humann_config --update database_folders protein タンパクデータベースの PATH
humann_config --update database_folders utility_mapping 代謝経路データベースの PATH
また、解析モードを run_modes で設定する。threads (スレット数)は高いほうが計算時間を短縮できる。verbose を True にしておくと解析の経過が表示されるため、処理落ちの確認などができる。一方で bypass_prescreen を True にすると prescreen が省略される。一連の解析をすべて実行したい場合、これらの bypass を True にする必要はない。
humann_config --update run_modes threads 12
humann_config --update run_modes verbose True
alignment_settings は解析結果に影響するパラメータである。現在調査中。
ショットガンメタゲノム解析の実行
次のコマンドで一連の解析を実行する。
humann -i INPUT_kneaddata.fastq -o result/
正規表現を用いた全ファイル一括実行は以下のようにするとよい。中間成果物フォルダ *_humann_temp/ から MetaPhlAn で得られた細菌群集構造データ *_metaphlan_bugs_list.tsv を取り出す。それ以外の中間成果物は不要であれば削除する。
for f in kneaddata_res/*_kneaddata.fastq;do humann -i $f -o humann_res/ ;\
mv humann_res/*_humann_temp/*_metaphlan_bugs_list.tsv humann_res/ ;\
rm -r humann_res/*_humann_temp/ ;done
最終成果物について
INPUT_kneaddata_genefamilies.tsv ==遺伝子ファミリーの存在量==
# Gene Family INPUT_kneaddata_Abundance-RPKs
UNMAPPED 4594940.0000000000
UniRef90_E4W393 284074.1793863305
UniRef90_E4W393|g__Bacteroides.s__Bacteroides_vulgatus 232823.9637420343
UniRef90_E4W393|g__Bacteroides.s__Bacteroides_fragilis 6641.9736016735
UniRef90_E4W393|g__Bacteroides.s__Bacteroides_xylanisolvens 6574.6010640810
UniRef90_E4W393|g__Bacteroides.s__Bacteroides_intestinalis 6461.5818734712
UniRef90_E4W393|g__Bacteroides.s__Bacteroides_uniformis 6345.4057487223
UniRef90_E4W393|g__Bacteroides.s__Bacteroides_thetaiotaomicron 6258.9343891602
UniRef90_E4W393|g__Bacteroides.s__Bacteroides_caccae 6223.1513834636
UniRef90_E4W393|g__Parabacteroides.s__Parabacteroides_distasonis 6216.8804594610
UniRef90_E4W393|unclassified 3399.4484659169
UniRef90_E4W393|g__Bacteroides.s__Bacteroides_ovatus 3128.2386583464
UniRef90_A0A015P9C8 218360.2201153699
UniRef90_A0A015P9C8|g__Bacteroides.s__Bacteroides_fragilis 218360.2201153699
- レコードの中身は
データベース名_遺伝子ファミリー名|g_属名.s_種名 PRKs(Read Per Kilobases)である。 - 各 gene family において、一行目は細菌群集全体がもつ存在量であり、二行目以降は各菌がもつ存在量である。
- 細菌群集全体の存在量は、各菌の存在量の総和に等しい。
INPUT_kneaddata_pathabundance.tsv ==代謝経路の存在量==
# Pathway INPUT_kneaddata_Abundance
UNMAPPED 1324989.4588691823
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast) 3567.4930431590
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_uniformis 964.5199427428
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Faecalibacterium.s__Faecalibacterium_prausnitzii 170.5596233500
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_xylanisolvens 118.7663621484
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_fragilis 44.9025217032
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_ovatus 35.5973187745
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|unclassified 28.4954735475
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_uniformis_CAG_3 11.3641690616
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis 3355.5531617011
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis|unclassified 168.9724785494
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis|g__Parabacteroides.s__Parabacteroides_merdae 89.0111890856
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis|g__Parabacteroides.s__Parabacteroides_distasonis 63.1827227249
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis|g__Lachnoclostridium.s__Clostridium_bolteae 39.2412978038
- レコードの中身は
Pathway ID: Pathway 名|g_属名.s_種名 PRKs(Read Per Kilobases)である。 - 各 pathway において、一行目は細菌群集全体がもつ存在量であり、二行目以降は各菌がもつ存在量である。
- 計算上、細菌群集全体の存在量が各菌の存在量の総和に等しいとは限らない。
INPUT_kneaddata_pathcoverage.tsv ==代謝経路のカバレッジ==
# Pathway INPUT_kneaddata_Coverage
UNMAPPED 1.0000000000
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast) 0.0000000000
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_uniformis 0.0965648630
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Faecalibacterium.s__Faecalibacterium_prausnitzii 0.0000000000
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_xylanisolvens 0.0000000000
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_fragilis 0.1127007775
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_ovatus 0.0000000000
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|unclassified 0.0000000000
PWY-7357: thiamin formation from pyrithiamine and oxythiamine (yeast)|g__Bacteroides.s__Bacteroides_uniformis_CAG_3 0.0000000000
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis 0.0000000000
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis|unclassified 0.0000523372
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis|g__Parabacteroides.s__Parabacteroides_merdae 0.5581117767
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis|g__Parabacteroides.s__Parabacteroides_distasonis 0.0845603346
COMPLETE-ARO-PWY: superpathway of aromatic amino acid biosynthesis|g__Lachnoclostridium.s__Clostridium_bolteae 0.0083868254
- レコードの中身は
Pathway ID: Pathway 名|g_属名.s_種名 coverageである。 - 各 pathway において、一行目は細菌群集全体の coverage であり、二行目以降は各菌の coverage である。
- Pathway abundance と比べてロバストな指標とされている。
重要な中間成果物について
INPUT_kneaddata_humann_temp/ フォルダに中間成果物が得られる。
INPUT_kneaddata_metaphlan_bugs_list.tsv ==各細菌の相対存在量==
# mpa_v30_CHOCOPhlAn_201901
# /Users/keisukeota/miniconda3/bin/metaphlan INPUT_kneaddata.fastq -t rel_ab -o INPUT_kneaddata_humann_temp/INPUT_kneaddata_metaphlan_bugs_list.tsv --input_type fastq --bowtie2out INPUT_kneaddata_humann_temp/INPUT_kneaddata_metaphlan_bowtie2.txt --nproc 12
# SampleID Metaphlan_Analysis
# clade_name NCBI_tax_id relative_abundance additional_species
k__Bacteria 2 100.0
k__Bacteria|p__Bacteroidetes 2|976 92.85956
k__Bacteria|p__Firmicutes 2|1239 6.8021
k__Bacteria|p__Proteobacteria 2|1224 0.33059
k__Bacteria|p__Verrucomicrobia 2|74201 0.00776
k__Bacteria|p__Bacteroidetes|c__Bacteroidia 2|976|200643 92.85956
k__Bacteria|p__Firmicutes|c__Clostridia 2|1239|186801 6.60863
k__Bacteria|p__Proteobacteria|c__Betaproteobacteria 2|1224|28216 0.29427
k__Bacteria|p__Firmicutes|c__Negativicutes 2|1239|909932 0.16878
k__Bacteria|p__Firmicutes|c__Firmicutes_unclassified 2|1239| 0.02468
k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria 2|1224|1236 0.02402
- 一行目は metaphlan コマンドが記載される。
- 二行目以降は各細菌(もしくはウイルス)の相対存在量が記載される。
- 細菌は
k__界(kingdom)|p__門(phylum)|c__網(class)|o__目(order)|g__属(genus)|s__種(species)で表示される。
成果物の統合
各サンプルから得られた HUMAnN3.0 の最終成果物は humann_join_tables で統合することができる。--file_name オプションは統合対象ファイルの名前の共通部分を入力する。
humann_join_tables -i humann_res/ -o humann_res/genefamilies.tsv --file_name genefamilies #遺伝子ファミリー
humann_join_tables -i humann_res/ -o humann_res/pathabundance.tsv --file_name pathabundance ## 代謝経路の存在量
humann_join_tables -i humann_res/ -o humann_res/pathcoverage.tsv --file_name pathcoverage ## 代謝経路のカバレッジ
正規化
Read 数が異なるサンプルを比較する際の正規化を genefamilies.tsv と pathabundance.tsv に対して実施する。
humann_renorm_table --input humann_res/genefamilies.tsv --units cpm --output humann_res/genefamilies_cpm.tsv ##遺伝子ファミリー
humann_renorm_table --input humann_res/pathabundance.tsv --units cpm --output humann_res/pathabundance_cpm.tsv ## 代謝経路の存在量