はじめに
Jupyter上で、PythonライブラリであるASE(Atomic Simulation Environment)を介して、第一原理計算ソフトQuantum Espressoで、第一原理計算を実施してみます。
本記事は、書籍『密度半関数理論入門(D.S.ショール,J.A.ステッケル 著、佐々木泰造,末原茂 共訳』(以下、教科書)の例題や課題を参考に、プログラムを作って計算します。
なお、固体DFTの専門家ではありませんので、計算条件、結果等はご自身でご確認/判断ください。
今回は、教科書2章の練習問題1、
『Ptが単純立方構造・fcc・hpcのいずれの構造をとるかをDFTから見積もる』
問題に取り組んでみたいと思います。
前準備
計算に必要になるライブラリや、擬ポテンシャルを用意します。
# import libs
import os
import subprocess
import numpy as np
import matplotlib.pyplot as plt
from scipy.optimize import curve_fit
# import ase
from ase.build import bulk
from ase.visualize import view
from ase.calculators.espresso import Espresso
#Cuの例にならい、Ptのpw91の擬ポテンシャルを使います。
!wget http://nninc.cnf.cornell.edu/psp_files/Pt.pw91-n-van.UPF
pp_file="Pt.pw91-n-van.UPF"
関数の定義
計算するための構造を作る関数、計算条件を設定する関数などを作ります。今回は結晶系や格子定数a,cを変量するので、それに対応した関数を作ります。
def make_crystal_lattice(type=None, a=None,c=None):
model = bulk(name='Pt', crystalstructure=type, a=a, c=c,orthorhombic=False, cubic=False, basis=None)
return model
def calc_dft_pt():
pseudopotentials = {'Pt':pp_file}
cmd = 'mpirun -np 5 pw.x -in espresso.pwi > espresso.pwo'
input_data = {
'control':{'pseudo_dir':'./'},
'system': {
'occupations' : 'smearing',
'smearing' : 'mp',
'degauss' : 0.001,
'ecutwfc': 25.0, #25[Ry] ~ 340[eV]
'ecutrho': 100.0}, #ecutwfc * 4
'disk_io': 'low'}
calc = Espresso(command=cmd,
pseudopotentials=pseudopotentials,
kpts=(12, 12, 12),
input_data=input_data)
return calc
def move_results_file(dir_for_result):
# QEの計算ファイルを別のフォルダにまとめて格納する
os.makedirs(dir_for_result, exist_ok=True)
subprocess.call('mv espresso.* pwscf.save pwscf.xml '+dir_for_result+'/.', shell=True)
subprocess.call('cp *.UPF '+dir_for_result+'/.', shell=True)
結晶系と格子定数を変えてDFT計算
まずは単純立方格子(sc)で格子定数aを変量してエネルギーを計算します。
l_type = 'sc'
a_list = np.arange(2.5, 3.05, 0.05)
r=[]
for lat_a in a_list:
print(f" a :{lat_a:.2f} [Angs.]")
Pt = make_crystal_lattice(type=l_type, a=lat_a)
Pt.set_calculator(calc_dft_pt())
r.append(Pt.get_potential_energy()/len(Pt))
move_results_file(l_type+"/a"+'{:.2f}'.format(lat_a))
a_sc = np.array(a_list)
r_sc = np.array(r)
###実行結果###
a :2.50 [Angs.]
a :2.55 [Angs.]
....
続いて面心立方格子fcc、六方最密構造hcpで格子定数を変えて計算します。hcpの方は、aとc/aを変量してエネルギーを計算します。計算数が多いこともあり、少し時間がかかります。できればcalc_dft_pt()内、cmdの-npの数字をPCのCPUコア数まで上げて計算してください。
# fcc
l_type = 'fcc'
a_list = np.arange(3.75, 4.3, 0.05)
r=[]
for lat_a in a_list:
print(f" a :{lat_a:.2f} [Angs.]")
Pt = make_crystal_lattice(type=l_type, a=lat_a)
Pt.set_calculator(calc_dft_pt())
r.append(Pt.get_potential_energy()/len(Pt))
move_results_file(l_type+"/a"+'{:.2f}'.format(lat_a))
a_fcc = np.array(a_list)
r_fcc = np.array(r)
#hcp
l_type = 'hcp'
c_per_a_list = np.arange(1.6, 1.85, 0.05) # For hcp #0.05
a_list = np.arange(2.5, 3.5, 0.05)
r_hcp=[]
for c_per_a in c_per_a_list :
r=[]
for lat_a in a_list :
print(f" c/a:{c_per_a:.2f}, a :{lat_a:.2f} [Angs.]")
Pt = make_crystal_lattice(type='hcp', a=lat_a, c=c_per_a*lat_a) # For hcp
Pt.set_calculator(calc_dft_pt())
r.append(Pt.get_potential_energy()/len(Pt))
fname=f'{l_type}/ca{c_per_a:.2f}_a{lat_a:.2f}'
move_results_file(fname)
r_hcp.append(r)
ca_hcp = np.array(c_per_a_list)
a_hcp = np.array(a_list)
r_hcp = np.array(r_hcp)
計算結果の解析
まずは、格子定数に対するエネルギー変化を見てみましょう。
# make a fig.
plt.rcParams["font.size"] = 12
fig = plt.figure(figsize=(5,4))
plt.xlabel("a [Angstrom]")
plt.ylabel("Etot/atom [eV]")
plt.plot(a_sc,r_sc,label="sc")
plt.plot(a_fcc,r_fcc,label="fcc")
for i, ca in enumerate(c_per_a_list):
plt.plot(a_hcp, r_hcp[i],label=f'hcp_ca{ca:.1f}')
plt.ylim(-1212.1,-1211.0)
plt.legend(fontsize=8)
plt.show()
fig.savefig("result.png")
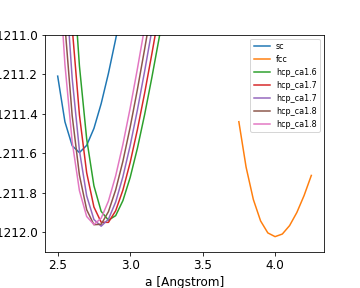
図 sc,fcc,hcpにおけるPtの格子定数aに対する全エネルギーEtot(a)
fccが若干エネルギーが低そうです。念のためそれぞれの構造のエネルギー最小値を確認してみましょう。
print(f'Emin(sc ) = {r_sc.min():.2f}')
print(f'Emin(fcc) = {r_fcc.min():.2f}')
print(f'Emin(hcp) = {r_hcp.min():.2f}')
print("----")
print(f'lattice_a @ Emin(fcc) = {a_fcc[r_fcc.argmin()]:.2f}')
###実行結果###
Emin(sc ) = -1211.60
Emin(fcc) = -1212.02
Emin(hcp) = -1211.97
----
lattice_a @ Emin(fcc) = 4.00
fccが最もエネルギーが低く、最適化された格子定数は4Åであることがわかります。
この値は、今回変量したaの中で最もエネルギーが低かったaということになります。
ここで、fccのDFT結果に、Birch Murnagham状態方程式にFittigしてa0と平衡体積弾性率B0を求めてみます。
def Birch_Murnagham_EoS(a, a0, B0, B02, E0):
#a:lattice_a
E = 9*(a**3)/4*B0/16 * ( B02*( (a0/a)**2-1)**3 + ( ((a0/a)**2-1)**2 * (6-4*(a0/a)**2) ) ) +E0
return E
print('Fit Birch Murnagham EoS')
init_param = np.array([4, 142, 0, 0]) #初期値与えないとうまく収束しない
fit_params, std = curve_fit(Birch_Murnagham_EoS, a_fcc, r_fcc, p0=init_param, maxfev=10000)
est_BMeos = Birch_Murnagham_EoS(a_fcc, *fit_params)
print(f' a0 = {fit_params[0]:.2f} [Angstrom]')
print(f' B0 = {fit_params[1]* 160.21766208:.2f} [GPa]')
###実行結果###
Fit Birch Murnagham EoS
a0 = 4.00 [Angstrom]
B0 = 244.71 [GPa]
a0 = 4.00 [Angstrom]、B0 = 244.71 [GPa]という結果。
実験値との比較
下記に他の資料からの値を掲載します。
- Crystal Structure: fcc (Spece Group : Fm-3m) [Ref.1]
- Lattice Constant : 3.925 [Angstrom] [Ref.1]
- Bulk modulus : 230[GPa] [Ref.2]
Ref.1 : Ebert H. et al, Z. Phys. Chem. v.144,p.223, (1985) by NIMS AtomWorks
Ref.2 : Wikipedia(実験値?)
DFTで求められた構造と格子定数はよく一致しているかと。B0は5%以上異なっていますがこんなものなんでしょうか....
ASEとQuantum Espresso関連の投稿記事リンク
ASEとQuantum Espressoを使って、
- 手動でCuの単位格子最適化して、平衡体積弾性率を見積る
- Ptが単純立方構造、fcc、hpcのいずれの構造をとるかを調べる
- 六方晶Hfの格子定数aとcを最適化する
- H2Oなどの3原子分子を構造を最適化して、安定構造を見つける
他