この記事では,LDA汎関数(Dirac-Slaterの交換汎関数(参考文献[1],参考文献[2])とVWN相関汎関数(参考文献[3])の組み合わせ)を用いて,水素原子に対するKohn-Sham方程式を解き,基底状態のエネルギーEを計算してみることにします。なお,Kohn-Sham方程式そのものについてはこちらの記事で,また,ヘリウム原子に対するKohn-Sham方程式については,こちらの記事で解説しています。
水素原子に対するKohn-Sham方程式
さて,早速ですが,水素原子に対するKohn-Sham方程式は,LDA-VWN汎関数を用いた場合,Hartree原子単位系で(以下,断りなしにHartree原子単位系を用いることとします),
\left[ -\dfrac{1}{2}\nabla^{2}-\dfrac{1}{r}+%
%TCIMACRO{\dint }%
%BeginExpansion
{\displaystyle\int}
%EndExpansion
\dfrac{n\left( \vec{r}^{\prime}\right) }{\left\vert \vec{r}-\vec{r}^{\prime
}\right\vert }d\vec{r}^{\prime}-\left( \dfrac{3}{\pi}\right) ^{\frac{1}{3}%
}n\left( \vec{r}\right) ^{\frac{1}{3}}+v_{c}^{VWN}\left( \vec{r}\right)
\right] \phi\left( \vec{r}\right) =\varepsilon\phi\left( \vec{r}\right) \quad(1)
となります。ただし,
n\left( \vec{r}\right) =\left\vert \phi\left( \vec{r}\right) \right\vert
^{2} \quad(2)
です。ここで,$ (1) $式の左辺括弧の中の第3項はHartreeポテンシャル,第4項はLDA交換ポテンシャル,左辺括弧の中の第5項はVWN相関ポテンシャルです。ここで,水素原子に対するSchrödinger方程式は,
\left( -\dfrac{1}{2}\nabla^{2}-\dfrac{1}{r}\right) \phi\left( \vec
{r}\right) =E\phi\left( \vec{r}\right) \quad(3)
でしたから,$ (1) $式と$ (3) $式を比較すると,$ (1) $式の右辺第3項のHartreeポテンシャル項と右辺第4項のLDA-VWN汎関数による交換ポテンシャル項はちょうど相殺し合って消え,また右辺第5項のLDA-VWN汎関数による相関ポテンシャル項はゼロにならなくてはならないことが分かります。すなわち水素原子の場合,右辺第3項のHartreeポテンシャル項は,その全てが自分自身と相互作用する非物理的な項ですが,これは右辺第4項の交換ポテンシャル項も同様であり,従ってこれらはちょうど相殺されなければなりません。また,右辺第5項の相関ポテンシャル項については,相関とはそもそも多体問題の効果(現実の系の運動エネルギー$ T $と,Kohn-Shamの補助系における運動エネルギー$ T_{s} $の差も含めて)でしたから,一体問題である水素原子に対しては,当然ながらゼロとならなければいけません。さらに,$ (1) $式の一電子軌道エネルギー$ \varepsilon $と,$ (3) $式のエネルギー$ E $は,完全に等しくならなければなりません。さて,LDA-VWN汎関数では,これらの条件は満たされているでしょうか。結論から言うと,LDA-VWN汎関数では,これらの条件は満たされません。これは,自己相互作用問題と呼ばれます。
さて,水素原子のエネルギーは,こちらの記事の$ (14) $式と、こちらの記事の$ (8) $式から,
から,
E=\varepsilon-\dfrac{1}{2}%
%TCIMACRO{\dint }%
%BeginExpansion
{\displaystyle\int}
%EndExpansion
\dfrac{n\left( \vec{r}\right) n\left( \vec{r}^{\prime}\right) }{\left\vert
\vec{r}-\vec{r}^{\prime}\right\vert }d\vec{r}d\vec{r}^{\prime}+%
%TCIMACRO{\dint }%
%BeginExpansion
{\displaystyle\int}
%EndExpansion
\left\{ -\dfrac{3}{4}\left( \dfrac{3}{\pi}\right) ^{\frac{1}{3}}n\left(
\vec{r}\right) ^{\frac{4}{3}}d\vec{r}+\epsilon_{c}^{VWN}\left( \left[
n\right] ,\vec{r}\right) \right\} n\left( \vec{r}\right) d\vec{r}-%
%TCIMACRO{\dint }%
%BeginExpansion
{\displaystyle\int}
%EndExpansion
\left[ -\left( \dfrac{3}{\pi}\right) ^{\frac{1}{3}}n\left( \vec{r}\right)
^{\frac{1}{3}}+v_{c}^{VWN}\left( \vec{r}\right) \right] n\left( \vec
{r}\right) d\vec{r} \quad(4)
となります。上述した,$ (1) $式の一電子軌道エネルギー$ \varepsilon $と,$ (3) $式のエネルギー$ E $は,完全に等しくならなければならない,という制限から,$ (4) $式の右辺第2項,第3項および第4項は,完全に相殺しなければならないという条件が課せられることになります。しかし,LDA-VWN汎関数は,この条件も満たしません。ここで,$ \epsilon_{c}^{VWN}\left( \left[ n\right] ,\vec{r}\right) $(あるいは$v_{c}^{VWN}(\vec{r}) $も)は,本来であれば二種類のスピン密度$ n^{\alpha}(\vec{r}) $および$ n^{\beta}(\vec{r}) $の両方に依存するのですが,水素原子の場合は,単に,
\begin{array}{ll}
n^{\alpha}\left( \vec{r}\right) =n\left( \vec{r}\right) \\
n^{\beta}\left( \vec{r}\right) =0
\end{array} \quad(5)
とすれば済みます。さてここで,この記事で行ったように,
\phi\left( \vec{r}\right) =%
%TCIMACRO{\dsum \limits_{p=1}^{n}}%
%BeginExpansion
{\displaystyle\sum\limits_{p=1}^{n}}
%EndExpansion
C_{p}\chi_{p}\left( \vec{r}\right) \quad(6)
なる$ n $個の基底を用いて,$ (1) $式を展開し,かつ左から$ \chi_{p}(\vec{r}) $を乗じ,$ \vec{r} $について全空間で積分すると,
F_{pq}=h_{pq}+%
%TCIMACRO{\dsum \limits_{r,s=1}^{n}}%
%BeginExpansion
{\displaystyle\sum\limits_{r,s=1}^{n}}
%EndExpansion
C_{r}C_{s}Q_{prqs}+K_{pq} \quad(7)
および
\mathbf{F}\vec{C}=\varepsilon\mathbf{S}\vec{C} \quad(8)
なる,一般化固有値問題が得られます。ここで,
h_{pq}=%
%TCIMACRO{\dint }%
%BeginExpansion
{\displaystyle\int}
%EndExpansion
d\vec{r}\chi_{p}\left( \vec{r}\right) \left( -\dfrac{1}{2}\nabla^{2}%
-\dfrac{1}{r}\right) \chi_{q}\left( \vec{r}\right) \quad(9) \\
Q_{prqs}=%
%TCIMACRO{\dint }%
%BeginExpansion
{\displaystyle\int}
%EndExpansion
d\vec{r}d\vec{r}^{\prime}\chi_{p}\left( \vec{r}\right) \chi_{r}\left(
\vec{r}^{\prime}\right) \dfrac{1}{\left\vert \vec{r}-\vec{r}^{\prime
}\right\vert }\chi_{q}\left( \vec{r}\right) \chi_{s}\left( \vec{r}^{\prime
}\right) \quad(10) \\
K_{pq}=%
%TCIMACRO{\dint }%
%BeginExpansion
{\displaystyle\int}
%EndExpansion
d\vec{r}\chi_{p}\left( \vec{r}\right) \left[ -\left( \dfrac{3}{\pi
}\right) ^{\frac{1}{3}}n\left( \vec{r}\right) ^{\frac{1}{3}}+v_{c}%
^{VWN}\left( \vec{r}\right) \right] \chi_{q}\left( \vec{r}\right) \quad(11) \\
S_{pq}=%
%TCIMACRO{\dint }%
%BeginExpansion
{\displaystyle\int}
%EndExpansion
d\vec{r}\chi_{p}\left( \vec{r}\right) \chi_{q}\left( \vec{r}\right) \quad(12)
です。なお,$ (9) $式は1電子積分,$ (10) $式はCoulomb積分,$ (12) $式は重なり積分と呼ばれます。また,$ (11) $式は,交換相関積分と呼ぶことにします。複雑そうに見えますが,やっていることは,この記事で解説したことと全く同じです)。後は$ (8) $式を数値的に解くだけです。
ここで,基底にGTOを用いると,基底状態については,$ l = 0 $の関数(s関数)だけが必要であり,これは,原子核に中心がある(そのためそこを原点に置く)ので,$ \chi_{p}(\vec{r}) $は,以下のようになります。
\chi_{p}(\vec{r}) =e^{-\alpha_{p}r^{2}} \quad(13)
$ (13) $式を用いると,水素原子における,1電子積分$ h_{pq} $とCoulomb積分$ Q_{prqs} $,および重なり積分$ S_{pq} $の要素は,解析的に求められ,次式のようになります(計算は省略します)。
h_{pq}=3\dfrac{\alpha_{p}\alpha_{q}\pi^{\frac{3}{2}}}{\left( \alpha
_{p}+\alpha_{q}\right) ^{\frac{5}{2}}}-\dfrac{2\pi}{\alpha_{p}+\alpha_{q}} \quad(14) \\
Q_{prqs}=\dfrac{2\pi^{\frac{5}{2}}}{\left( \alpha_{p}+\alpha_{q}\right)
\left( \alpha_{r}+\alpha_{s}\right) \sqrt{\alpha_{p}+\alpha_{q}+\alpha
_{r}+\alpha_{s}}} \quad(15) \\
S_{pq}=\left( \dfrac{\pi}{\alpha_{p}+\alpha_{q}}\right) ^{\frac{3}{2}} \quad(16)
ただし,$ (11) $式の交換相関積分$ K_{pq} $だけは,以下の式によって,数値的に求めなければなりません。
K_{pq}=4\pi%
%TCIMACRO{\dint _{0}^{\infty}}%
%BeginExpansion
{\displaystyle\int_{0}^{\infty}}
%EndExpansion
r^{2}\chi_{p}\left( r\right) \left[ -\left( \dfrac{3}{\pi}\right)
^{\frac{1}{3}}n\left( r\right) ^{\frac{1}{3}}+v_{c}^{VWN}\left( r\right)
\right] \chi_{q}\left( r\right) dr \quad(17)
ここで,具体的な$ \alpha_{p} $の値についてですが,参考文献[4]に,$ n = 4 $の場合($ \phi\left(\vec{r}\right) $を4つのGTOで展開した場合)の値が記載されているので,それをそのまま引用します(なお一般に$ \alpha_{p} $の値は,非線形の最適化問題を解くことによって求められますが,ここではその方法については立ち入りません)。その値は,
\begin{array}{ll}
\alpha_{1}=0.1219492 \\
\alpha_{2}=0.444529 \\
\alpha_{3}=1.962079 \\
\alpha_{4}=13.000773
\end{array} \qquad(18)
です。さてこれで,行列要素を埋めるための情報が全て得られましたので,以下の手続きに従って,$ (8) $式を自己無撞着に解くだけです。
- 固有ベクトル$ \vec{C} $の要素$ C_{p} $の初期値を適当に選びます。これは例えば,全部$ 1 $とかで構いません(今は水素原子という単純な系を考えているので,たいていの初期値から正しい解に収束するはずです。ただ,全部0だけはダメです)。また,重なり行列$ \mathbf{S} $も計算しておきます。
- 固有ベクトル$ \vec{C} $を,
\vec{C}:=\dfrac{1}{\sqrt{4\pi%
%TCIMACRO{\dsum \limits_{p=1}^{n}}%
%BeginExpansion
{\displaystyle\sum\limits_{p=1}^{n}}
%EndExpansion%
%TCIMACRO{\dsum \limits_{q=1}^{n}}%
%BeginExpansion
{\displaystyle\sum\limits_{q=1}^{n}}
%EndExpansion
C_{p}C_{q}%
%TCIMACRO{\dint _{0}^{\infty}}%
%BeginExpansion
{\displaystyle\int_{0}^{\infty}}
%EndExpansion
r^{2}\chi_{p}\left( r\right) \chi_{q}\left( r\right) dr}}\vec{C} \quad(19)
とすることにより,Kohn-Sham固有関数を正規化します。
3. 電子密度$ n(r) $を,
n\left( r\right) =\left[
%TCIMACRO{\dsum \limits_{p=1}^{n}}%
%BeginExpansion
{\displaystyle\sum\limits_{p=1}^{n}}
%EndExpansion
C_{p}\chi_{p}\left( r\right) \right] ^{2} \quad(20)
により求めます。
4. ステップ2で求めた固有ベクトル$ \vec{C} $と,ステップ3で求めた電子密度$ n(r) $から,$ (7) $式によって,Fock行列$ \mathbf{F} $を計算します。
5. $ (8) $式の一般化固有値問題を数値的に解きます。
6. エネルギー$ E $を,
E=\varepsilon-\dfrac{1}{2}%
%TCIMACRO{\dsum \limits_{p,q,r,s=1}^{m}}%
%BeginExpansion
{\displaystyle\sum\limits_{p,q,r,s=1}^{m}}
%EndExpansion
C_{p}C_{q}C_{r}C_{s}Q_{prqs}+K^{\prime}-%
%TCIMACRO{\dsum \limits_{p,q=1}^{n}}%
%BeginExpansion
{\displaystyle\sum\limits_{p,q=1}^{n}}
%EndExpansion
K_{pq} \quad(21)
により求めます。ここで,$ K^{\prime} $は,
K^{\prime}=4\pi%
%TCIMACRO{\dint _{0}^{\infty}}%
%BeginExpansion
{\displaystyle\int_{0}^{\infty}}
%EndExpansion
r^{2}\left[ -\dfrac{3}{4}\left( \dfrac{3}{\pi}\right) ^{\frac{1}{3}%
}n\left( \vec{r}\right) ^{\frac{4}{3}}d\vec{r}+\epsilon_{c}^{VWN}\left(
\left[ n\right] ,\vec{r}\right) \right] n\left( r\right) dr \quad(22)
で定義されますが,これは数値的に求めなければなりません。
7. ステップ5で求まった固有ベクトル$ \vec{C} $から,電子密度$ n(r) $を$ (20) $式によって求め,これらから,Fock行列$ \mathbf{F} $を計算します。
8. ステップ5~7を,前回計算したエネルギー$ E_{old} $と,今回計算したエネルギー$ E_{new} $の差の絶対値$ \left\vert E_{new}-E_{old}\right\vert $が,ある閾値未満になるまで繰り返します。
LDA-VWN汎関数によってヘリウム原子のエネルギーを求めるプログラム
上述の方法によって,ヘリウム原子の(基底状態の)エネルギーを求める,C++17のプログラムは以下のようになります(多次元配列のためにBoost C++ Librariesを,また一般化固有値問題の解を求めるためにEigenを,また交換相関エネルギーおよび交換相関ポテンシャルの計算のためにLibxcを,そして数値積分のためにALGLIBをそれぞれ使っていますので,注意してください)。ただし,全部載せると非常に長くなってしまうので,一部だけを掲載しています。なお,完全なコードはGitHubのこちらで公開しています。
std::optional<double> Hydrogen_LDA::do_scfloop()
{
// GTOの肩の係数が格納された配列を生成
make_alpha();
// 1電子積分が格納された2次元配列を生成
make_oneelectroninteg();
// 2電子積分が格納された4次元配列を生成
make_twoelectroninteg();
// 重なり行列を生成
make_overlapmatrix();
// 全て1.0で初期化された固有ベクトルを生成
make_c(1.0);
// 固有ベクトルを正規化
normalize();
// 新しく計算されたエネルギー
auto enew = 0.0;
// SCFループ
for (auto iter = 1; iter < Hydrogen_LDA::MAXITER; iter++) {
// Fock行列を生成
make_fockmatrix();
// 一般化固有値問題を解く
Eigen::GeneralizedSelfAdjointEigenSolver<Eigen::MatrixXd> es(f_, s_);
// εを取得
epsilon = es.eigenvalues()[0];
// 固有ベクトルを取得
c_ = es.eigenvectors().col(0);
// 前回のSCF計算のエネルギーを保管
auto const eold = enew;
// 今回のSCF計算のエネルギーを計算する
enew = calc_energy();
std::cout << boost::format("Iteration # %2d: KS eigenvalue = %.14f, energy = %.14f\n") % iter % epsilon % enew;
// SCF計算が収束したかどうか
if (std::fabs(enew - eold) < Hydrogen_LDA::SCFTHRESHOLD) {
// 収束したのでそのエネルギーを返す
return std::make_optional(enew);
}
}
// SCF計算が収束しなかった
return std::nullopt;
}
double Hydrogen_LDA::calc_energy()
{
// E = ε
auto e = epsilon;
for (auto p = 0; p < nalpha_; p++) {
for (auto q = 0; q < nalpha_; q++) {
for (auto r = 0; r < nalpha_; r++) {
for (auto s = 0; s < nalpha_; s++) {
// E -= 0.5 * ΣCp * Cq * Cr * Cs * Qprqs
e -= 0.5 * c_[p] * c_[q] * c_[r] * c_[s] * q_[p][q][r][s];
}
}
}
}
// E += K'
e += calc_exc_energy();
for (auto p = 0; p < nalpha_; p++) {
for (auto q = 0; q < nalpha_; q++) {
// E -= ΣCp * Cq * Kpq
e -= c_[p] * c_[q] * k_[p][q];
}
}
return e;
}
double Hydrogen_LDA::calc_exc_energy() const
{
using namespace boost::math::constants;
auto const func = [this](double x)
{
auto rhotemp = 0.0;
for (auto r = 0; r < nalpha_; r++)
{
rhotemp += c_[r] * std::exp(-alpha_[r] * x * x);
}
rhotemp *= rhotemp;
// 電子密度
std::array<double, 2> rho = { rhotemp, 0.0 };
// 交換相関エネルギー
std::array<double, 2> zk_x{}, zk_c{};
// 交換エネルギーを求める
xc_lda_exc(pxfunc_.get(), 1, rho.data(), zk_x.data());
// 相関エネルギーを求める
xc_lda_exc(pcfunc_.get(), 1, rho.data(), zk_c.data());
return x * x * (zk_x[0] + zk_c[0]) * rhotemp;
};
// K'を求める
return 4.0 * pi<double>() * gl_.qgauss(func, 0.0, Hydrogen_LDA::MAXR);
}
void Hydrogen_LDA::input_nalpha()
{
while (true) {
std::cout << "使用するGTOの個数を入力してください (3, 4 or 6): ";
std::cin >> nalpha_;
if (!std::cin.fail() && (nalpha_ == 3 || nalpha_ == 4 || nalpha_ == 6)) {
break;
}
std::cin.clear();
std::cin.ignore(Hydrogen_LDA::MAXBUFSIZE, '\n');
}
}
void Hydrogen_LDA::make_alpha()
{
switch (nalpha_) {
case 3:
alpha_ = { 0.16885539999999999, 0.62391373000000006, 3.4252509099999999 };
break;
case 4:
alpha_ = { 0.1219492, 0.444529, 1.962079, 13.00773 };
break;
case 6:
alpha_ = { 0.100112428, 0.24307674700000001, 0.62595526599999995, 1.8221429039999999, 6.5131437249999999, 35.523221220000003 };
break;
default:
BOOST_ASSERT(!"make_alpha()関数のswitch文のdefaultに来てしまった!");
break;
}
}
void Hydrogen_LDA::make_c(double val)
{
c_.resize(nalpha_);
// 固有ベクトルCの要素を全てvalで初期化
for (auto i = 0; i < nalpha_; i++) {
c_[i] = val;
}
}
void Hydrogen_LDA::make_exchcorrinteg()
{
using namespace boost::math::constants;
for (auto p = 0; p < nalpha_; p++) {
for (auto q = 0; q < nalpha_; q++) {
auto const func =[this, p, q](double x)
{
auto rhotemp = 0.0;
for (auto r = 0; r < nalpha_; r++)
{
rhotemp += c_[r] * std::exp(-alpha_[r] * x * x);
}
rhotemp *= rhotemp;
// 電子密度
std::array<double, 2> rho = { rhotemp, 0.0 };
// 交換相関ポテンシャル
std::array<double, 2> zk_x{}, zk_c{};
// 交換ポテンシャルを求める
xc_lda_vxc(pxfunc_.get(), 1, rho.data(), zk_x.data());
// 相関ポテンシャルを求める
xc_lda_vxc(pcfunc_.get(), 1, rho.data(), zk_c.data());
return x * x * std::exp(-alpha_[p] * x * x) * (zk_x[0] + zk_c[0]) * std::exp(-alpha_[q] * x * x);
};
// Kpqの要素を埋める
k_[p][q] = 4.0 * pi<double>() * gl_.qgauss(func, 0.0, Hydrogen_LDA::MAXR);
}
}
}
void Hydrogen_LDA::make_fockmatrix()
{
// 交換相関積分を計算
make_exchcorrinteg();
for (auto p = 0; p < nalpha_; p++) {
for (auto qi = 0; qi < nalpha_; qi++) {
// Fpq = hpq + ΣCr * Cs * Qprqs + Kpq
f_(p, qi) = h_[p][qi] + k_[p][qi];
for (auto r = 0; r < nalpha_; r++) {
for (auto s = 0; s < nalpha_; s++) {
f_(p, qi) += c_[r] * c_[s] * q_[p][r][qi][s];
}
}
}
}
}
void Hydrogen_LDA::make_oneelectroninteg()
{
using namespace boost::math::constants;
for (auto p = 0; p < nalpha_; p++) {
for (auto q = 0; q < nalpha_; q++) {
// αp + αq
auto const appaq = alpha_[p] + alpha_[q];
// hpq = 3αpαqπ^1.5 / (αp + αq)^2.5 - 2π / (αp + αq)
h_[p][q] = 3.0 * alpha_[p] * alpha_[q] * std::pow((pi<double>() / appaq), 1.5) / appaq -
2.0 * pi<double>() / appaq;
}
}
}
void Hydrogen_LDA::make_overlapmatrix()
{
using namespace boost::math::constants;
for (auto p = 0; p < nalpha_; p++) {
for (auto q = 0; q < nalpha_; q++) {
// Spq = (π / (αp + αq))^1.5
s_(p, q) = std::pow((pi<double>() / (alpha_[p] + alpha_[q])), 1.5);
}
}
}
void Hydrogen_LDA::make_twoelectroninteg()
{
using namespace boost::math::constants;
for (auto p = 0; p < nalpha_; p++) {
for (auto qi = 0; qi < nalpha_; qi++) {
for (auto r = 0; r < nalpha_; r++) {
for (auto s = 0; s < nalpha_; s++) {
// Qprqs = 2π^2.5 / [(αp + αq)(αr + αs)√(αp + αq + αr + αs)]
q_[p][r][qi][s] = 2.0 * std::pow(pi<double>(), 2.5) /
((alpha_[p] + alpha_[qi]) * (alpha_[r] + alpha_[s]) *
std::sqrt(alpha_[p] + alpha_[qi] + alpha_[r] + alpha_[s]));
}
}
}
}
}
void Hydrogen_LDA::normalize()
{
using namespace boost::math::constants;
auto sum = 0.0;
for (auto p = 0; p < nalpha_; p++) {
for (auto q = 0; q < nalpha_; q++) {
sum += c_[p] * c_[q] / (4.0 * (alpha_[p] + alpha_[q])) * std::sqrt(pi<double>() / (alpha_[p] + alpha_[q]));
}
}
for (auto p = 0; p < nalpha_; p++) {
c_[p] /= std::sqrt(4.0 * pi<double>() * sum);
}
}
なお,このプログラムは,参考サイト[1]のhelium_hf_gauss.cを参考にさせて頂きました。また,$ n = 3 $および$ n = 6 $の場合の$ \alpha_{p} $の値は,参考サイト[2]の,basis-sto3g.cppおよびbasis-sto6g.cppから引用させて頂きました。
プログラムの実行結果
前節で紹介したプログラムを実行した結果は,以下の画像のようになります。
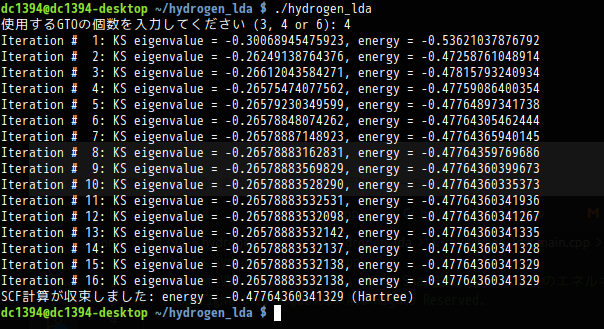
$ n = 4 $で,固有ベクトル$ \vec{C} $の要素$ C_{p} $の初期値がすべて1の場合,$ \left\vert E_{new}-E_{old}\right\vert $が,閾値(今の場合$ 10^{-15} $)未満になるまでに,16回のループを要していることが分かります。
プログラムによる水素原子のエネルギーの計算結果の結果と考察
ここで,このプログラムによる$ n = 3 $,$ n = 4 $,$ n = 6 $のときのエネルギーの計算結果を表に示しますと,以下のようになります(単位は全てHartree原子単位です)。
| n | Kohn-Sham (LDA-VWN) | 通常の基底による展開 |
|---|---|---|
| 3 | -0.4728 | -0.4957 |
| 4 | -0.4776 | -0.4993 |
| 6 | -0.4784 | -0.4999 |
| 無限個の基底を用いた極限 | −0.4787 | −0.5000 |
| 厳密 | −0.5000 | −0.5000 |
上記の表を見ると,LDA-VWN汎関数を用いたKohn-Sham法による計算結果は,$ n = 3 $,$ n = 4 $,$ n = 6 $,また無限個の基底による極限(この値は,有限要素法によって高精度にKohn-Sham方程式を解く拙作のコードの計算結果を用いました)の場合のいずれも,水素原子に対する通常のSchrödinger方程式を,単に基底で展開した場合の計算結果に比べて劣っていることが分かります。これは一体なぜなのでしょうか(ただし,劣っていると言っても,厳密な計算結果と比較して,4.3%の過小評価(無限個の基底を用いた極限の場合)であり,厳密な計算結果と比較して,54%も過大評価するThomas-Fermiモデルより,遙かに良くなっていることには注意すべきです)。結論から言うと,この原因は,上述した自己相互作用にあります。
LDA-VWN汎関数による水素原子の電子密度,Thomas-Fermiモデルによる電子密度および厳密な電子密度の比較
では,水素原子におけるLDA-VWN汎関数による電子密度,Thomas-Fermiモデルによる電子密度および厳密な電子密度を,プロットで比較してみることにしましょう(なお,この章のLDA-VWN汎関数による計算には,上述の拙作のコードを用いました)。
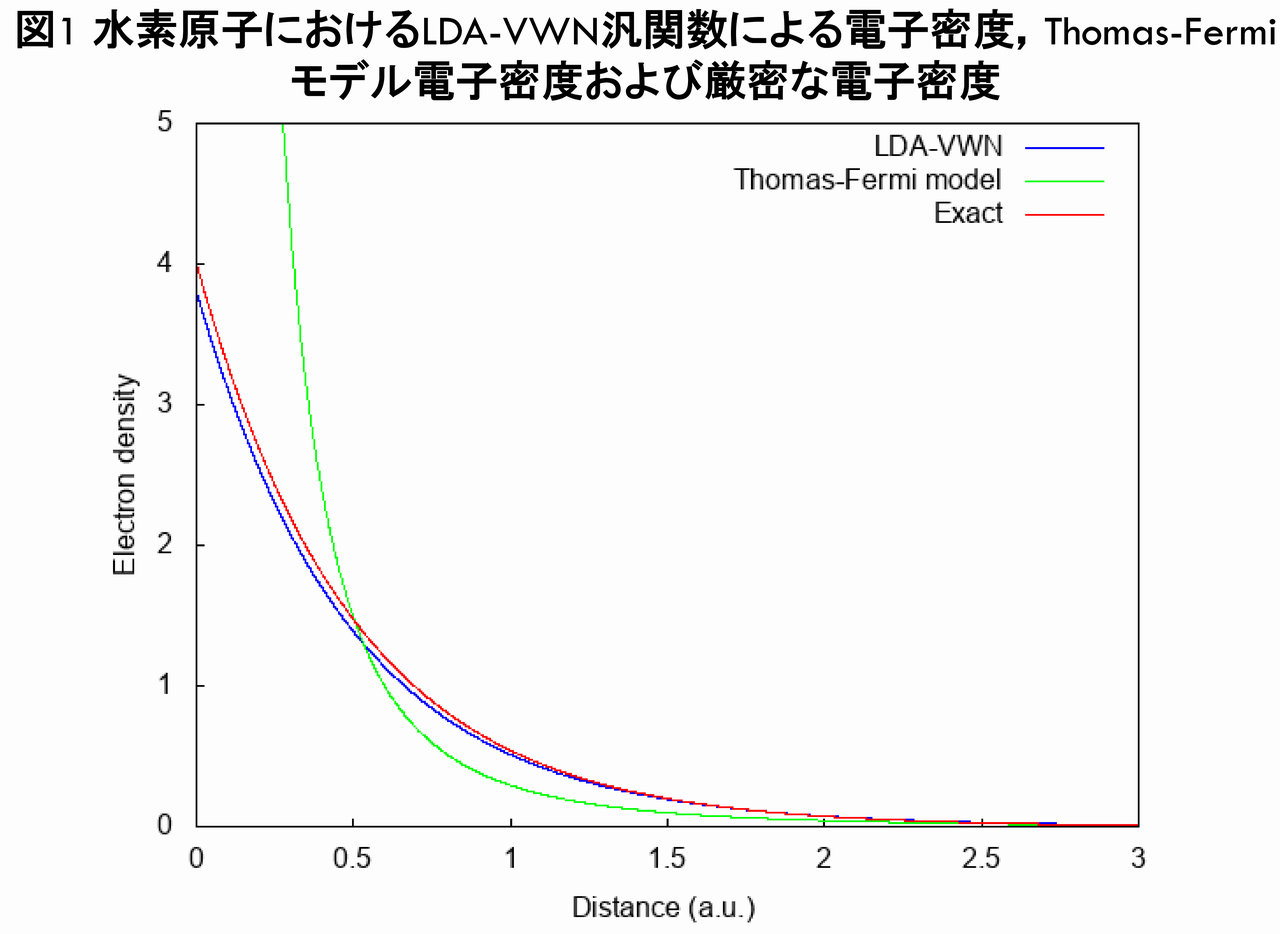
ここで,青線は,LDA-VWN汎関数による電子密度であり,緑線はThomas-Fermiモデルの電子密度,赤線は厳密な電子密度です。図1から分かることは,LDA-VWN汎関数による電子密度は,Thomas-Fermiモデルの電子密度より,厳密な電子密度に遙かに近くなっているということです。実際,LDA-VWN汎関数による電子密度は,Thomas-Fermiモデルの電子密度のように,原点で発散していません。これは,Thomas-Fermiモデルからの大きな進歩だと言えます。
次に,y軸を対数目盛にした場合のグラフを示します。
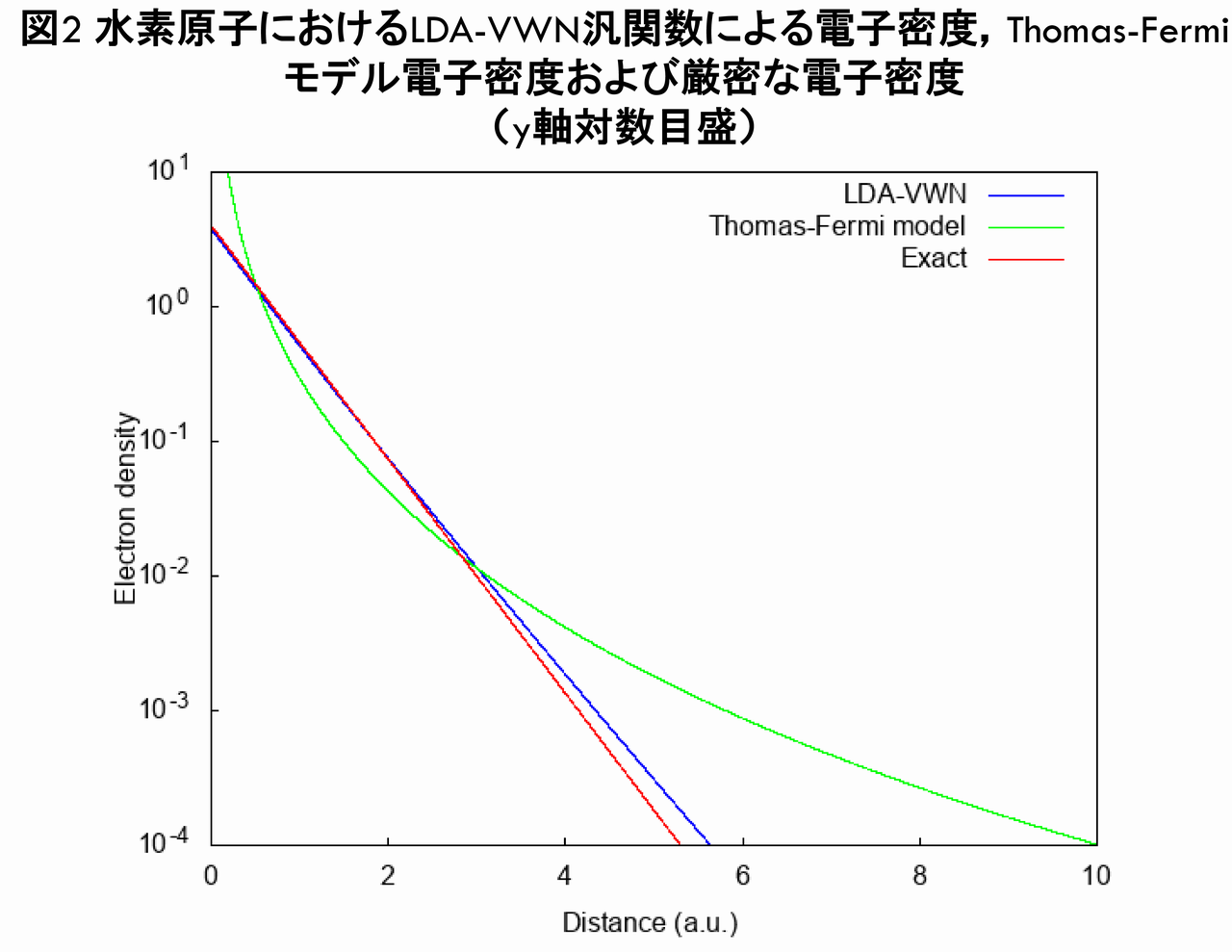
図2からは,LDA-VWN汎関数による電子密度は,Thomas-Fermiモデルの電子密度とは異なり,厳密な電子密度のように指数関数で減衰していることが分かります。さらに,動径方向の電荷分布(電子密度に$ r^{2} $を乗じたもの)を比較したグラフが下図です。
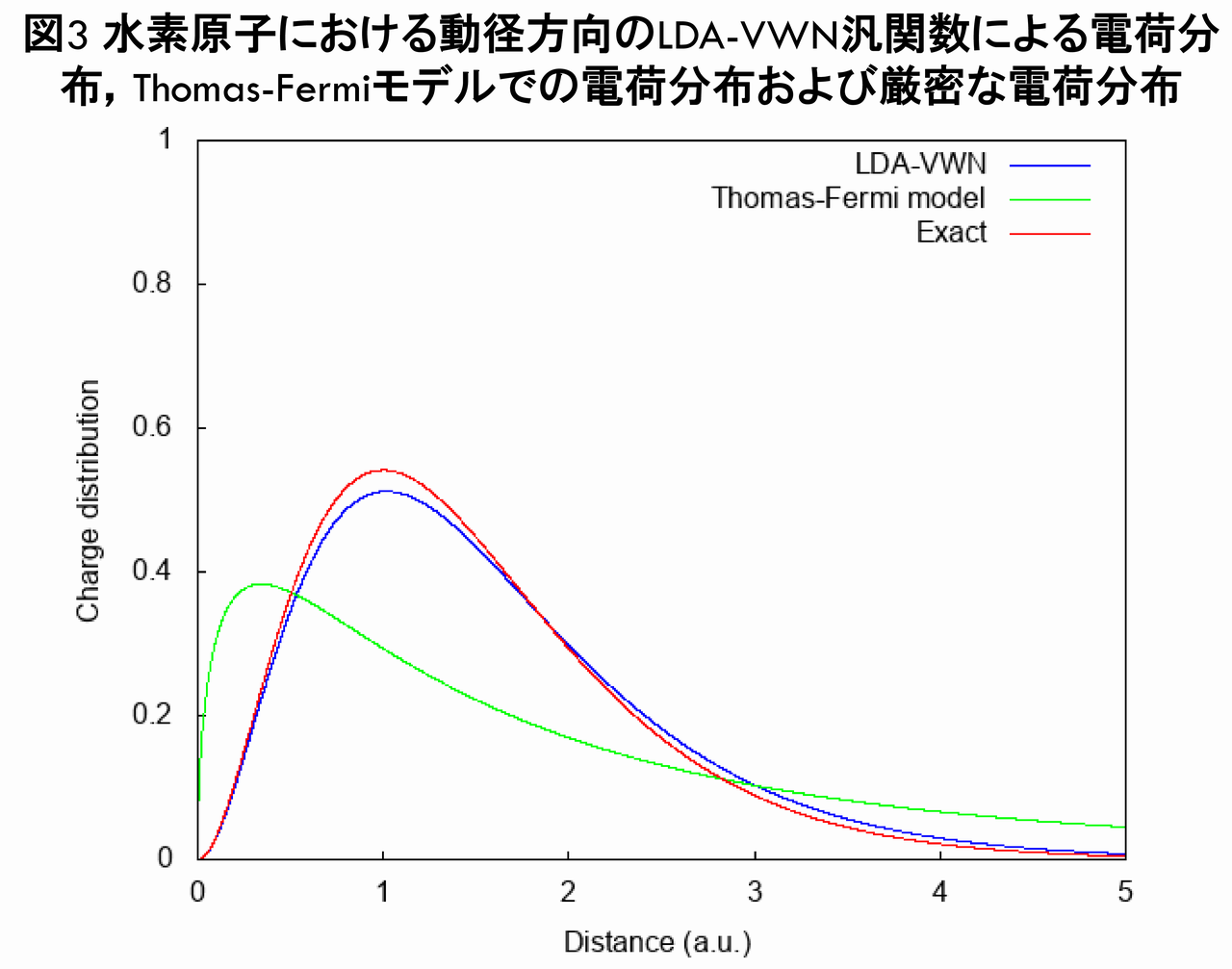
図3から分かるとおり,LDA-VWN汎関数による電子密度は,厳密な電子密度に良く似ていることが分かります。また,この電荷分布を三次元プロットで比較したのが下図です。

図4から分かるとおり,LDA-VWN汎関数の電荷分布の様子は,図5の厳密な電荷分布の様子を再現していることが分かります(この記事の図5と比べてみてください。なお,電荷分布の三次元プロットには拙作のコードを用いました)。
最後に
上述のように,LDA-VWN汎関数を用いた計算結果は,Thomas-Fermiモデルの計算結果より,遙かに優れたものとなっていますが,厳密な計算結果にはまだ及びません。この原因は,既に述べたように自己相互作用にあります。自己相互作用問題はKohn-Sham法における大問題であり,また別の記事で詳しく論じたいと思います。
参考文献
[1] P. A. M. Dirac, (Mathematical) Proceedings of the Cambridge Philosophical Society 26, 376 (1930).
[2] J. C. Slater, Phys. Rev. 81(3), 385 (1951).
doi:10.1103/PhysRev.81.385
[3] S. H. Vosko, L. Wilk, and M. Nusair, Can. J. Phys. 58, 1200 (1980).
doi:10.1139/p80-159
[4] J.M.ティッセン 『計算物理学』シュプリンガー・フェアラーク東京(2003)