Transcriptdbについて
GenomicFeaturesパッケージのTranscriptDbオブジェクトでtranscriptのメタデータを管理することができる.TranscriptDbオブジェクトの裏にはSQLiteデータベースがある.
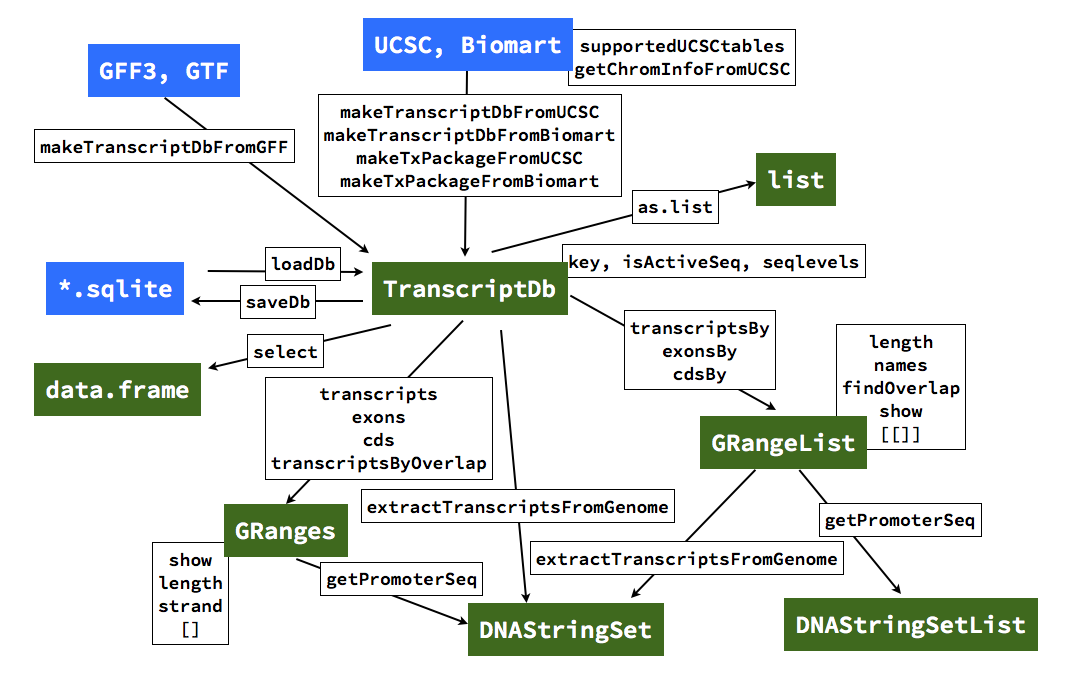
TranscriotDbオブジェクトと周辺のクラスとの関係
パッケージの読み込み
## source("http://bioconductor.org/biocLite.R")
## biocLite("GenomicFeatures")
library("GenomicFeatures")
TranscriptDbオブジェクトのロード
元々あるデータからTranscriptDbオブジェクトをロードするには2つの方法があります.一つはloadDBを使う方法で,.sqliteファイルを直接読み込みます.もう一つはパッケージとして提供されているものを使う方法です.
# Load transcript DB
samplefile <- system.file("extdata", "UCSC_knownGene_sample.sqlite",
package="GenomicFeatures")
txdb <- loadDb(samplefile)
txdb
# Load as annotation package
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
ls("package:TxDb.Hsapiens.UCSC.hg19.knownGene") #check object name
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene #shorthand (for convenience)
txdb
TrancsriotDbオブジェクト
is(txdb)
metadata(txdb)
seqinfo(txdb)
head(seqinfo(txdb)@seqlengths)
seqlengths(txdb)
seqlevels(txdb)
names(which(isCircular(txdb)))
# Use of as.list
txdump <- as.list(txdb)
is(txdump)
names(txdump)
## txdump
txdb1 <- do.call(makeTranscriptDb, txdump) stopifnot(identical(as.list(txdb1), txdump))
データを染色体でフィルタする
# See which chromosomes are currently available
isActiveSeq(txdb)
# List of chromosomes and/or contigs
seqlevels(txdb)
# Filter by chromosome
isActiveSeq(txdb)[seqlevels(txdb)] <- FALSE
# same as: isActiveSeq(txdb) <- rep(FALSE, length(seqinfo(txdb)))
isActiveSeq(txdb) <- c("chr15"=TRUE)
selectでSQLのようにデータをデータフレームとして抽出できる
# Which keys are available
keytypes(txdb)
# Which values are available
cols(txdb)
keys <- c("100033416", "100033417", "100033420")
selected <- select(txdb, keys=keys, cols=c("TXNAME", "TXSTRAND"), keytype="GENEID")
selected
is(selected)
区間をGRangesオブジェクトとして取得する
GR <- transcripts(txdb)
is(GR)
GR
GR[1:3]
length(GR)
# Filter by vals
GR <- transcripts(txdb, vals <- list(tx_chrom = "chr15", tx_strand = "+"))
length(GR)
unique(strand(GR))
# Extract Exons as ranges
EX <- exons(txdb)
EX[1:4]
length(EX)
length(GR)
TSSの座標(幅1bpの区間)を抽出する
(gr <- GR[3956:3959])
(extractedTSS <- resize(gr, width=1, fix="start"))
is(extractedTSS)
ある区間と重なるものを区間として抽出する
GR <- GRanges(seqnames = rep("chr15",2),
ranges = IRanges(start=c(500,70000000), end=c(10000,75000000)), strand = strand(rep("-",2)))
(extractedGR <- transcriptsByOverlaps(txdb, GR))
is(extractedGR)
区間を何らかのまとまりでまとめて(byで指定),GRangesListオブジェクトとして取得する
GRList <- transcriptsBy(txdb, by = "gene")
length(GRList)
names(GRList)[10:13]
GRList[11:12]
is(GRList)
#
GRList <- exonsBy(txdb, by = "tx")
length(GRList)
# Elements of GRangesList is accessed by [[]]
GRList[[12]]
tx_ids <- names(GRList)
head(select(txdb, keys=tx_ids, cols="TXNAME", keytype="TXID"))
# More specific
length(intronsByTranscript(txdb))
length(fiveUTRsByTranscript(txdb))
length(threeUTRsByTranscript(txdb))
TranscriptDbで指定されている塩基配列をDNAStringSetとして取り出す
library(BSgenome.Hsapiens.UCSC.hg19)
ls("package:BSgenome.Hsapiens.UCSC.hg19")
is(Hsapiens)
# Extract transcripts from genome
tx_seqs1 <- extractTranscriptsFromGenome(Hsapiens, txdb)
is(tx_seqs1)
length(tx_seqs1)
tx_seqs2 <- extractTranscriptsFromGenome(Hsapiens, exonsBy(txdb,by="tx"))
is(tx_seqs2)
length(tx_seqs2)
# CDS
cds_seqs <- extractTranscriptsFromGenome(Hsapiens, cdsBy(txdb, by="tx"))
translate(cds_seqs)
# Promoter sequences
## GRanges --> DNAStringSet
promoter_seqs1 <- getPromoterSeq(transcripts(txdb), Hsapiens, upstream=10, downstream=0)
is(promoter_seqs1)
length(promoter_seqs1)
## GRangesList --> DNAStringSetList
promoter_seqs2 <- getPromoterSeq(exonsBy(txdb,by="tx"), Hsapiens, upstream=10, downstream=0)
is(promoter_seqs2)
length(promoter_seqs2)
TranscriptDbオブジェクトをつくる,パッケージ化する
# Which tables in UCSC are available
supportedUCSCtables()
## mm9KG <- makeTranscriptDbFromUCSC(genome = "mm9", tablename = "knownGene")
head(getChromInfoFromUCSC("hg19"))
## mmusculusEnsembl <-
## makeTranscriptDbFromBiomart(biomart = "ensembl",
## dataset = "mmusculus_gene_ensembl")
TranscriptDbオブジェクトを保存する
## saveDb(mm9KG, file="fileName.sqlite")
## mm9KG <- loadDb("fileName.sqlite")
SessionInfo
sessionInfo()
Sys.time()