(おことわり)
本記事は初心者が試行錯誤した過程を備忘録を兼ねてまとめたものです。
不正確な記述などありましたらご指摘いただけますと幸いです。
はじめに
pymolの製品版はここからDLできる。
https://www.pymol.org/
使い方はTogoTVでも紹介されている。
しかし、製品版の教育版ライセンスでは、出版目的での利用は禁止されている。
そこで、オープンソース版をインストールして表面電荷表示などのプラグインを導入することを試みる。
Anacondaインストール→Anacondaでpymolをインストール
ここでは、minicondaを使用
Anaconda install から pymol installまでのやりかた解説はコチラ
おそらくこれが一番簡単。
ここまでで、pymolの使用自体はできるようになる。カンタン!
(参考)Anacondaインストール後からのみの解説
(参考)conda install を使わない方法
(おまけ)ショートカット用のバッチファイルを作成
以下のコードを"pymol.bat"として保存
echo off
cd C:\Users\USERNAME\miniconda3
cmd.exe /K "Scripts\activate.bat base && conda activate pymol && pymol"
(出典)
これで、batファイルをダブルクリックすれば開くようにはなった。
ただ、bat ファイルはスタートメニューにピン止めできないらしい。
を参考に、ショートカットを作成→プロパティからリンクをいじる→できたファイルをタスクバーやスタートメニューに追加
とやることで、ボタン一つで起動できるようになる!
ここから、Anaconda環境にapbsをインストールしていく
次に、以下の画像のように表面電荷を表示できるAPBSをインストールする。
Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, Chun M, Li P, Gohara DW, Dolinsky T, Konecny R, Koes DR, Nielsen JE, Head-Gordon T, Geng W, Krasny R, Wei G-W, Holst MJ, McCammon JA, Baker NA. Improvements to the APBS biomolecular solvation software suite. Protein Sci, 27 (1), 112-128, 2018. DOI:10.1002/pro.3280
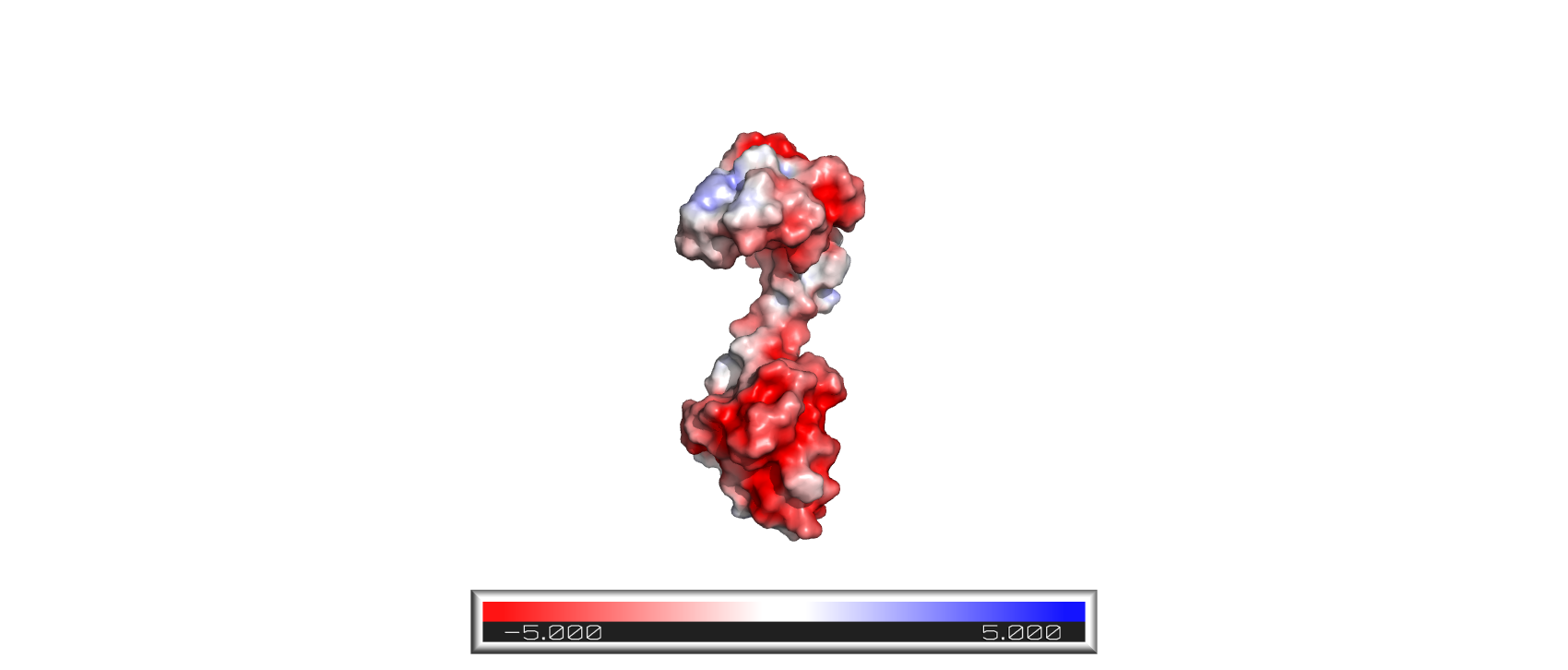
(PDB ID: 1cll)
※環境の確認
conda create -n pymol # 仮想環境の構築
conda activate pymol # 仮想環境の起動
conda install -c conda-forge pymol-open-source
を行った、という前提で進める。
(ちなみに、執筆時にはpymol_scという別環境を構築し、そこに製品版をインストールして環境を照らし合わせながら作業していた。時たま出てくるpymol_scはそういう意味です。)
Anaconda Pronptにて
conda activate pymol # 仮想環境の起動
conda install conda-forge::apbs
conda install conda-forge::pdb2pqr
(出典)
何か聞かれたら、yで進める
インストールに成功すると、pluginタブから"Run"でプログラムを実行できるはず…
なのだが…うまくいかなかったので、対処法を以下にまとめる。
結論から言うと、以下のように正しい.exeファイルを指定できれば動く。
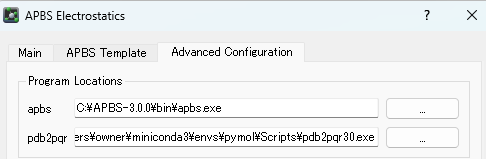
しかし、初期設定ではプログラムファイルの指定先が正しくない場合がある。
簡単な場合だと、以下のように同じディレクトリ内にある.exeファイルを指定するとうまくいくと思われる。
C:\Users\username\miniconda3\envs\pymol\Scripts\pdb2pqr
↓
C:\Users\username\miniconda3\envs\pymol\Scripts\pdb2pqr30.exe
一方、apbs.exeはというと…
製品版では miniconda3\envs\pymol_sc\Library\bin
にあるのだが、open source ver. にはない。
インストール画面を見ると、インストールに失敗しているようである(2024.11.29時点)
(この段階で成功すれば、この先は必要ないのかもしれない。)
(pymol) C:\Users\owner>conda install conda-forge::apbs
Channels:
- defaults
- conda-forge
Platform: win-64
Collecting package metadata (repodata.json): done
Solving environment: failed
PackagesNotFoundError: The following packages are not available from current channels:
- conda-forge::apbs
Current channels:
- defaults
- https://conda.anaconda.org/conda-forge
To search for alternate channels that may provide the conda package you're
looking for, navigate to
https://anaconda.org
and use the search bar at the top of the page.
APBSのWebsiteではgithubに行き、DLするように案内されている
ここからだとwindows版のソースコードをDLできる。
インストールした後、自分でコンパイルする必要がある…?(よくわかっていません…)
上のサイトのAPBS3.0、あるいは以下のサイトからコンパイル済みのZipファイルが入手できるようである(2024.11.30時点)。
とりあえず筆者は以下のサイトからDLしてうまくいった。
いずれにせよ、何とかしてapbs.exeを入手できれば良い。そのファイルの置き場所を指定してあげて、以下のような状態になれば完了!
パラメーター
ここには何を入れるの?という話
pdb2pqrのサイトで、各項目にカーソルを合わせると出てくるコマンドを使えばよい。
(一応メモ。上記サイトを見る方がわかりやすい。)
# select foce field & pH control
--ff=AMBER
--titration-state-method=propka --with-ph=7
ex:
--ff=PARSE --titration-state-method=propka --with-ph=7
PARSEについて…
For most applications, the choice is easy: PARSE. This forcefield has been optimized for implicit solvent calculation and is probably the best choice for visualization of biomolecular electrostatics and many common types of energetic calculations for biomolecules. However, AMBER and CHARMM may be more appropriate if you are attempting to compare directly to simulations performed with those force fields, require nucleic acid support, are simulating ligands parameterized with those force fields, etc.
とのこと。
# options
Ensure that new atoms are not rebuilt too close to existing atoms: --nodebump と打たない
Optimize the hydrogen bonding network: --noopt と打たない
Assign charges to the ligand specified in a MOL2 file: --noopt
Create an APBS input file: --apbs-input=FILENAME.in
Add/keep chain IDs in the PQR file: --keep-chain
Insert whitespaces between atom name and residue name, between x and y, and between y and z: --whitespace
Make the protein's N-terminus neutral (requires PARSE forcefield): --neutraln
Make the protein's C-terminus neutral (requires PARSE forcefield): --neutralc
Remove the waters from the output file: --drop-water
AMBERではpH2でもC末端がプロトン化されなかったので、
--ff=PARSE --titration-state-method=propka --with-ph=2 --neutralc
とする必要がありそう。
初期設定の書き換え
ここまでの作業で、使用自体は可能。
ただこのままだと、開くたびに実行ファイルを選びなおさなくてはいけないのでめんどくさい。
ファイルの置き場所や書き換え方が異なっていたので以下にメモ。
" C:\Users\owner\miniconda3\envs\pymol\Lib\site-packages\pymol\pymol_path\data\startup\apbs_gui_init_.py "
のファイルを、以下のように書き換える。
# executables # 変更前
from shutil import which
form.apbs_exe.setText(electrostatics.find_apbs_exe() or 'apbs')
form.pdb2pqr_exe.setText(
which('pdb2pqr') or
which('pdb2pqr30') or
# acellera::htmd-pdb2pqr provides pdb2pqr_cli
which('pdb2pqr_cli') or 'pdb2pqr')
# executables # 変更後
from shutil import which
form.apbs_exe.setText(which(R"C:\APBS-3.0.0\bin\apbs.exe") or electrostatics.find_apbs_exe() or 'apbs')
form.pdb2pqr_exe.setText(
which(R"C:\Users\owner\miniconda3\envs\pymol\Scripts\pdb2pqr30.exe") or
which('pdb2pqr') or
which('pdb2pqr30') or
# acellera::htmd-pdb2pqr provides pdb2pqr_cli
which('pdb2pqr_cli') or 'pdb2pqr')
数点コメント
・ディレクトリパスを指定する""の前にRをつけること
・find_executableではなくwhichを使用
以下、おまけ
参考にしたサイトたち
pymol の使い方
森脇先生のページにかなりよくまとめられている。一方で環境構築の記事はmac向け。
本記事でやっていることのフローとしてはこれ。
また、疎水性度を表示するだけならこちらの記事を参照。
自分用メモ
・仮想環境の名前
pymol オープンソース版
pymol_sc 製品版
・製品版のデフォルトのパス
apbs
C:\Users\username\miniconda3\envs\pymol_sc\Library\bin\apbs.EXE
C:\Users\username\AppData\Local\Schrodinger\PyMOL2\Library\bin\apbs.EXE
pdb2pqr
C:\Users\username\miniconda3\envs\pymol_sc\Scripts\pdb2pqr #これだと走らない
C:\Users\username\AppData\Local\Schrodinger\PyMOL2\Scripts\pdb2pqr.EXE
.exeファイルを指定する!