はじめに
なるべく自分自身の力でまとめ上げましたが、私の憶測や偏見が一部あるかと思います。怪しい箇所は一次文献を参照してください。
目次
- 背景
- 目的
- 方法
- 内容
- どの分子記述子が良いのか?
- 所感
背景
化学を機械学習に適応させるには、化合物の数値化が必須です。その数値化は分子記述子という方法で行われます。この分子記述子はいくつも開発されており、私の中で整理できていませんでした。
目的
主な分子記述子について違いを整理すること。
それらをrdkitで実装し、すぐに機械学習で使える形に整理すること。
方法
Molecular descriptor (Wikipedia)の情報から派生して体系的に整理する。
pythonの実行環境は以下の通りである。
- python 3.7.5
- rdkit 2020.09.1.0
- mordred 1.2.0
内容
分子記述子の全体像を説明した後、pythonでの実装結果を説明します。
分子記述子
分子記述子 (Molecular Descriptor)は、実験実測値と理論分子記述子に大別されます。
さらに理論分子記述子は次元によって分類されます。次元による分類については、やや曖昧な部分があります。例えば、分子の表面積のような2次元の特性だけを説明変数とする場合は、化学スペースという空間では1次元と言えます。ここでは特性に関してn次元という表記に統一します。
分子記述子
┣ 実験実測値 (普遍特性)
┃ ┗ log P、屈折率、双極子モーメント、極性、原子のラベル付け、分子量、NMRシフト etc.
┃
┗ 理論分子記述子
┣ 0次元
┃ ┗ 原子タイプ、結合様式 etc.
┣ 1次元
┃ ┗ 原子タイプの数、水素結合ドナーやアクセプターの数、環の数、タイプ別の官能基の数、フィンガープリント etc.
┣ 2次元
┃ ┗ グラフ理論や計算された値による数学的表現 etc.
┣ 3次元
┃ ┗ 幾何学的記述子、極性表面積、3D-MoRSE記述子、WHIM記述子、GETAWAY記述子 etc.
┗ 4次元
┗ GRID法、CoMFA法、Volsurf etc.
実験実測値 (Experimental Measurements)
実験実測値というものの、rdkitで出力されるのは予測値です。本来は実験して実測することで、一意の値に決定されるという意味で実験実測値という言葉になっているのだと思います。
from rdkit import Chem
from rdkit.Chem import Crippen
from rdkit.Chem import Descriptors
mol = Chem.MolFromSmiles('CC(=O)Oc1ccccc1C(=O)') # アスピリンのsmilesからMolオブジェクトの生成
Crippen.MolLogP(mol) # log P:1.4244
Crippen.MolMR(mol) # 屈折率:43.13850000000002
Descriptors.MolWt(mol) # 分子量:164.15999999999997
理論分子記述子 (Theoretical Molecular Descriptors)
0次元
化合物を図形として捉えると、原子は点であり結合は線です。これらは0次元に分類され、単体では重要な情報を持たないですが、これらを組み合わせることで有用な情報を形成します。
from rdkit import Chem
mol = Chem.MolFromSmiles('CC(=O)Oc1ccccc1C(=O)') # アスピリンのsmilesからMolオブジェクトの生成
atom = mol.GetAtomWithIdx(0) # Atomオブジェクトの生成
atom.GetSymbol() # 元素記号の取得:C
bond = mol.GetBondWithIdx(0) # Bondオブジェクトの生成
bond.GetBondType() # 結合タイプの取得:rdkit.Chem.rdchem.BondType.SINGLE
1次元
分子記述子の代名詞と言ってもいいほど有名なフィンガープリント (Fingerprint)は1次元に分類され、Structure keysとHashed fingerprintsの2種類があります。フィンガープリントは計算速度と精度のバランスからよく使われるため、主なものについて詳しく説明します。
| Structure keys | Hashed fingerprints | |
| 特徴 | 予め部分構造の辞書を作成しておき、それに該当する構造があれば1を、なければ0のビットを立てる手法 | ある大きさよりも大きくないサイズのフラグメントを全て列挙して、それらをハッシュ関数を用いて0 or 1のビットに変換する手法 |
| 欠点 | 予め定義していない部分構造は捉えることはできない | ハッシュ化する際に、異なる構造同士が同じビットに割り当てられる「衝突」が起こって情報が欠落する |
フィンガープリント
┣ Structure keys
┃ ┗ MACCS keys
┃
┣ Hashed fingerprints
┃ ┃
┃ ┣ Path-based fingerprints
┃ ┃ ┗ RDKit fingerprint
┃ ┃
┃ ┗ Circular fingerprints
┃ ┣ ECFP
┃ ┣ FCFP
┃ ┣ MHFP
┃ ┣ Avalon
┃ ┗ Atom pair fingerprint
┃
┗ その他のフィンガープリント
┣ rdkitのDescriptorsモジュール
┗ mordred
【MACCS (Molecular ACCess System) keys】
MACCS keysはStructure keysであり、166ビットの部分構造情報 (表示上は167だが、1ビットは情報保持に利用される)を示します。どのような部分構造に基づいているのかは元コードを参照してください。コメント部分を流し読みするだけでもどんな部分構造を設定しているのかが何となく分かります。
from rdkit import Chem
mol1 = Chem.MolFromSmiles('C=CCCCCl')
mol2 = Chem.MolFromSmiles('C=CC(Cl)CC')
mols = [mol1, mol2]
for i,mol in enumerate(mols):
macc_fp = Chem.MACCSkeys.GenMACCSKeys(mol)
macc_fp = np.array(macc_fp, int)
print(i)
print(len(macc_fp))
print(macc_fp)
"""
0
167
[0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 1 0 0 0 0 0 0 0
0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 1
0 0 0 0 0 1 0 1 0 0 0 0 0 0 0 0 0 0 0]
1
167
[0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 1 0 0 0 1 0 0 0
0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 1 0 0 0 0 1 0 0 0 0 0 0]
"""
【RDKit fingerprint】
rdkitに詳細な記載はなかったのですが、予め設定しておいた結合長さ分の部分構造を列挙し、それに対してビットを立てていると思われます。アルゴリズムは公式HPを参考に説明します。長さがminPath (デフォルト1)とmaxPath (デフォルト7)の間のすべてのサブグラフ (部分構造)を見つけることによって機能します。
サブグラフごとに
- 結合タイプと各結合の隣接数を使用して、パスのハッシュを生成します
- ハッシュを使って乱数ジェネレーターをシードします
- 0 から fpSize までの nBitsPerHash 乱数を生成し、対応するビットを設定します
後述する「rdkitのDescriptorsモジュール」と混同しないように注意です。RDKit fingerprintは2048ビット情報を有しており、Daylightフィンガープリント (Hashed fingerprintでは有名)に類似したものです。
from rdkit import Chem
mol1 = Chem.MolFromSmiles('C=CCCCCl')
mol2 = Chem.MolFromSmiles('C=CC(Cl)CC')
mols = [mol1, mol2]
for i,mol in enumerate(mols):
rdkit_fp = Chem.RDKFingerprint(mol)
rdkit_fp = np.array(rdkit_fp, int)
print(i)
print(len(rdkit_fp))
print(rdkit_fp)
"""
0
2048
[0 0 0 ... 0 0 0]
1
2048
[0 0 0 ... 0 0 0]
"""
【ECFP (Extended Connectivity Fingerprint)】
ECFPは分子中の各原子の近傍の部分構造を探索して数値化します。ECFP4は直径4つまり、注目する原子の半径radius=2の範囲で部分構造を探索します。radius=2はGetMorganFingerprintAsBitVect()の引数に対応しています。
ECFPの詳細について知りたい方はココをクリック。
文献に従って、ECFPの概要を説明します。ECFPは4つのステージに分けられます。
- 原子識別子の初期割り当て
- 識別子の繰り返し更新
- 重複構造の除去
- 重複識別子の削除
【原子識別子の初期割り当て】
n個の原子で構成されている分子の各原子に番号 (1~n)をつけ、各原子の6つのDaylightの原子不変数を算出する。
- すぐ隣の重原子 (水素以外の原子)の数
- 水素を引いた原子価
- 原子番号
- 原子量
- 原子電荷
- 結合している水素の数
- 原子が少なくとも1つの環内にあるか否か
※ 7.はEFCPでのみ追加
これらをハッシュ関数で32bitの整数値に変換し、初期原子識別子とする。

【識別子の繰り返し更新】
注目している原子のarrayを以下のように用意する。ここではカルボニル基の根本 (4番)を例に上げる。
[1, -1100000244]
→ 1は繰り返しレベルを示し、はじめは1。
→ -1100000244は前項で計算した初期原子識別子である。
周りの原子の情報をこのarrayに加える。
[1, 初期原子識別子]
→ 1は結合様式。1,2,3,4は単結合、二重結合、三重結合、芳香結合に対応する。
→ 初期原子識別子は周りの原子情報。
→ これをソートして加える。ソートは結合様式、識別子の値の順で行う。
今回の例では、以下のようにarrayは更新される。
[1,-1100000244, 1, 1559650422, 1, 1572579716, 2,-1074141656]
このarrayをハッシュ化して、新しい一つの値になる。これらを予め決めておいた回数だけ繰り返す。
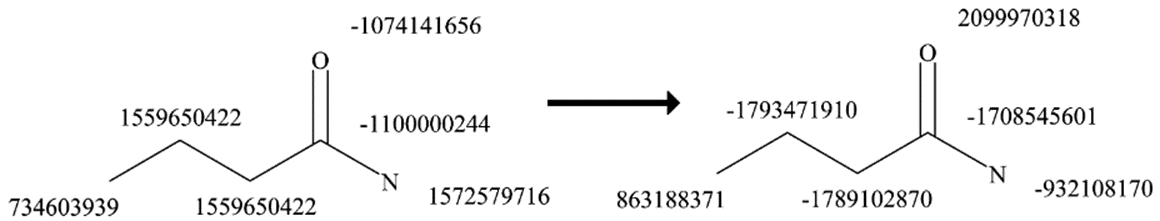
【重複構造の除去】
下図のアミド結合のように、OもしくはNを中心にしても、同じ範囲の情報を取得していることになる。これを構造重複という。ハッシュ化された値は異なっていたとしても、フィンガープリントに冗長性を与えないために、これは除去される必要がある。

除去するために、フィンガープリント特性は結合を表すセットを追跡する。
繰り返しから新たに生成された特徴がフィンガープリントに加えられる前に、いかなる構造重複が前回の特徴もしくは新たに生成された特徴に存在しないかをチェックされる。
構造重複が発見された場合は以下のように除去される。
- 異なる繰り返し番号なら、大きい繰り返し番号からの特徴を削除する。
- 同じ繰り返し番号なら、ハッシュ値が大きい方を削除する。
【重複識別子の削除】
重複識別子は等価の部分構造が分子中で1回以上現れるときに起こる。例えば、Me基が分子内でいくつも見られる場合などである。この重複識別子は削除する場合もあるし、残す場合もあるが、著書中では削除する。置換基を数えるフィンガープリントなら残す。文献には具体的な削除方法の記載はなし。
【ハッシュ関数の選択】
ここまでハッシュ関数について具体的な説明をしなかったが、合理的なハッシュ関数ならどれでも使用できる。重要なことは、ハッシュ関数が整数の配列をランダムかつ均一に2^32サイズの空間にマッピングすることである。均一にカバーしないと、衝突率が増加し、情報が失われる可能性がある。64bitを使用すれば、2^64サイズ空間が与えられ、衝突率はより下げられるが、64bitでは測定上の改善は確認されず、計算速度が低下した。
from rdkit.Chem import AllChem
import numpy as np
from rdkit import Chem
mol1 = Chem.MolFromSmiles('C=CCCCCl')
mol2 = Chem.MolFromSmiles('C=CC(Cl)CC')
mols = [mol1, mol2]
for i,mol in enumerate(mols):
ecfp_fp = AllChem.GetMorganFingerprintAsBitVect(mol, radius=2, nBits=1024)
ecfp_fp = np.array(ecfp_fp, int)
print(i)
print(len(ecfp_fp))
print(ecfp_fp)
"""
0
1024
[0 0 0 ... 0 0 0]
1
1024
[0 1 0 ... 0 0 0]
"""
【FCFP (Functional Connectivity Fingerprint)】
FCFPはECFPと似たような仕組みで、分子中の各原子の指定した半径内の情報を計算します。
実装方法もECFPと類似しています。GetMorganFingerprintAsBitVect()の引数useFeaturesをTrueにするだけです。
FCFPの詳細について知りたい方はココをクリック。初めにECFPの詳細をご覧になった方が分かりやすいです。
【イントロダクション】
前述のECFPの計算では不都合な場合もある。
ECFPでは、ベンゼン環上のClとBrはECFPでは全く異なるものとして扱われるが、置換反応の求電子剤という面では、どちらも類似した構造と捉えることができる。他にも、水素アクセプターは同じタイプとして扱いたいと考えるかもしれない。そこで、より抽象的に分子を取り扱えるFCFPが導入される。
【概要】
各原子は6bitで識別化される。
- 水素アクセプターorドナー
- + or – イオン
- 芳香環か否か
- ハロゲンか否か
一度識別子が計算されれば、後はECFPと処理は同じである。初期の識別化がECFPのと比較して、抽象的で結合情報も原子情報も環情報も周辺の原子情報も持たないため、同じハッシュ値が複数の構造を示すことがある。逆に言うと、類似化合物を検索したい時に、漏れが少ない。
from rdkit.Chem import AllChem
import numpy as np
from rdkit import Chem
mol1 = Chem.MolFromSmiles('C=CCCCCl')
mol2 = Chem.MolFromSmiles('C=CC(Cl)CC')
mols = [mol1, mol2]
for mol in mols:
fcfp = AllChem.GetMorganFingerprintAsBitVect(mol, radius=2, nBits=1024, useFeatures=True)
print(np.array(fcfp, int))
print(len(fcfp))
"""
[1 0 0 ... 0 0 0]
1024
[1 0 0 ... 0 0 0]
1024
"""
【MHFP (MinHash FingerPring)】
元文献の情報から解説します。MHFPは、ECFPの問題点 (上手く機能させるには高次元にする必要があり、ビッグデータの類似検索には時間がかかること)から起因して開発されました。
MHFPは、各原子から指定された範囲内の部分構造をSMILESで表現し、重複を除いて分子シングリングS(A)というリストにします。S(A)をMinHashというハッシュ関数にかけたいのですが、SMILESを直接適応させることはできないので、32bitに変換するハッシュ関数にかけます。そして、これをMinHashにかけることで最終的なフィンガープリントを得ます。
私は数学に親しくなく、ハッシュ関数やMinHashの部分は正直よく分かりませんでした。解説できる方がいらっしゃいましたらコメント欄までお願いします。

import numpy as np
from rdkit.Chem import rdMHFPFingerprint
mol1 = Chem.MolFromSmiles('C=CCCCCl')
mol2 = Chem.MolFromSmiles('C=CC(Cl)CC')
mols = [mol1, mol2]
encoder = rdMHFPFingerprint.MHFPEncoder()
for mol in mols:
mhfp = encoder.EncodeMol(mol)
print(len(np.array(mhfp)))
print(np.array(mhfp))
"""
2048
[224392011 172778395 173086079 ... 332130884 249896246 139349792]
2048
[764635273 9537623 268312112 ... 258922278 271720006 509648460]
"""
【Avalon】
ネットで調査しましたが、中々ヒットせず、ノバルティスが発表していた資料しか参考になりませんでした。以下の図は資料からの抜粋であり、図中右上の文献は有料で読めませんでしたが、QSARについての文献で著者がノバルティスの方らしいです。下図を手掛かりに読み解くと、ヘテロ環や水素の数、環についての特性をピックアップしていました。ノバルティスは製薬企業なので、類似性評価というよりもQSARの目的で作成した可能性が高いです。

from rdkit.Avalon import pyAvalonTools
from rdkit import Chem
mol1 = Chem.MolFromSmiles('C=CCCCCl')
mol2 = Chem.MolFromSmiles('C=CC(Cl)CC')
mols = [mol1, mol2]
for i,mol in enumerate(mols):
avalon_fp = pyAvalonTools.GetAvalonFP(mol)
avalon_fp = np.array(avalon_fp)
print(i)
print(avalon_fp)
print(len(avalon_fp))
"""
0
[0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0
0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 1
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 1 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 0 0 1 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 1 0 0 0 0 0 0 0
0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0]
512
1
[0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 0 0 0
0 0 0 0 1 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0
1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 1
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0
0 0 1 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 0 0 1 1 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0]
512
"""
【Atom Pair fingerprint】
分子内の全ての原子ペアを算出し、各原子の種類と最短距離と結合様式からキーを作成し、それをビット配列とします。局所的な特徴を抽出するCircular Fingerprintと異なり、遠く離れた原子間の情報を取得できることから、分子全体の構造を捉えることができます (元文献)。しかしながら、原子間の最短距離から特徴を作成するため、置換基の位置関係によっては位置異性体が同じフィンガープリントになってしまう場合があります。下図では同じフィンガープリントになることが確認されています。

from rdkit import Chem
from rdkit.Chem.AtomPairs import Pairs
mol1 = Chem.MolFromSmiles('C=CCCCCl')
mol2 = Chem.MolFromSmiles('C=CC(Cl)CC')
mols = [mol1, mol2]
for i,mol in enumerate(mols):
atompair_fp = Pairs.GetAtomPairFingerprintAsBitVect(mol)
atompair_fp = np.array(atompair_fp)
print(i)
print(atompair_fp)
print(len(atompair_fp))
"""
0
[0 0 0 ... 0 0 0]
8388608
1
[0 0 0 ... 0 0 0]
8388608
"""
【rdkitのDescriptorsモジュール】
rdkitのDescriptorsモジュールは部分構造の数や実験実測値を記述子として含みます。これまで説明してきたものとは異なり、0や1以外の数字が登場します。中身は記述子の名前と関数のタプルになっており、以下のコードで確認することができます。
from rdkit.Chem import Descriptors
print(len(Descriptors._descList))
print()
print('item\tfunction')
for i in Descriptors._descList[:5]:
print('{}\t{}'.format(i[0], i[1]))
'''
208
item function
MaxEStateIndex <function MaxEStateIndex at 0x00000227DBB65438>
MinEStateIndex <function MinEStateIndex at 0x00000227DBB654C8>
MaxAbsEStateIndex <function MaxAbsEStateIndex at 0x00000227DBB65558>
MinAbsEStateIndex <function MinAbsEStateIndex at 0x00000227DBB655E8>
qed <function qed at 0x00000227DBBA0DC8>
'''
実際に使用する際は、以下のような辞書を作成してpandasのデータフレームに落とし込んで、機械学習モデルに適応することになると思います。
def rtn_rdkit_desc(mol):
dic = {}
for tup in Descriptors._descList:
dic[tup[0]] = tup[1](mol)
return dic
mol = Chem.MolFromSmiles('CC(=O)Oc1ccccc1C(=O)')
rtn_rdkit_desc(mol)
"""
{'MaxEStateIndex': 5.531635802469135,
'MinEStateIndex': 0.17129629629629606,
'MaxAbsEStateIndex': 5.531635802469135,
'MinAbsEStateIndex': 0.17129629629629606,
'qed': 0.3722373316057521,
'MolWt': 104.58,
...}
"""
【mordred】
mordredはrdkitのDescriptorsモジュールと同様に、0や1以外の数字を含む記述子を提供します。約1800ビットの情報を有しており、2次元特性を約90%、3次元特性を約10%含んでいます (mordredの記述子一覧)。3次元特性を記述できますが、予め立体配座を最適化しておく必要があり、信頼できる立体配座を用意できない場合はignore_3D=Trueにしておくのが無難かと思います。mordredの性能比較は文献で行われています。
import pandas as pd
from mordred import Calculator, descriptors
mol1 = Chem.MolFromSmiles('C=CCCCCl')
mol2 = Chem.MolFromSmiles('C=CC(Cl)CC')
mols = [mol1, mol2]
calc=Calculator(descriptors, ignore_3D=False)
df = pd.DataFrame({'MOL':mols})
md=calc.pandas(df.loc[:, "MOL"])
df_mrg = pd.merge(df,md, left_index=True, right_index=True)
print(df_mrg.shape)
df_mrg
"""
(2, 1827)
MOL ABC ABCGG nAcid nBase SpAbs_A SpMax_A SpDiam_A SpAD_A SpMAD_A ... SRW10 TSRW10 MW AMW WPath WPol Zagreb1 Zagreb2 mZagreb1 mZagreb2
0 <rdkit.Chem.rdchem.Mol object at 0x000001188BE... 3.535534 3.869735 0 0 6.987918 1.801938 3.603875 6.987918 1.164653 ... 6.608001 28.105124 104.039278 6.935952 35 3 18.0 16.0 3.000000 1.750000
1 <rdkit.Chem.rdchem.Mol object at 0x000001188C1... 3.644924 4.097495 0 0 6.898979 1.931852 3.863703 6.898979 1.149830 ... 7.280008 29.753427 104.039278 6.935952 31 4 20.0 19.0 3.611111 1.666667
"""
2次元
説明のために以下の化合物を例に上げて説明します。
図のように原子の近くに番号を振る方法はここをクリック
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole, rdMolDraw2D
from IPython.display import SVG
m = Chem.MolFromSmiles('C=CC(Cl)CC')
view = rdMolDraw2D.MolDraw2DSVG(300, 300)
option = view.drawOptions()
option.addAtomIndices=True
view.DrawMolecule(m)
view.FinishDrawing()
svg = view.GetDrawingText()
SVG(svg)

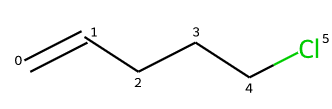
【隣接行列】
n個の原子で構成されている分子について、各原子に結合している原子の数をn×nの行列で示します。多くのトポロジカル指数 (接続の様式を数値化した指標)は隣接行列をベースに考えられています (文献)。
from rdkit import Chem
mol = Chem.MolFromSmiles('C=CC(Cl)CC')
print(Chem.GetAdjacencyMatrix(mol))
"""
[[0 1 0 0 0 0]
[1 0 1 0 0 0]
[0 1 0 1 1 0]
[0 0 1 0 0 0]
[0 0 1 0 0 1]
[0 0 0 0 1 0]]
"""
上のアウトプットでは分かりにくいので、テーブル形式にしました。2行目を見ると、1,3,4列目に1が入力されており、これは上図の2番の原子に1,3,4番の原子が結合していることに対応しています。
| 0 | 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|---|
| 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 1 | 1 | 0 | 1 | 0 | 0 | 0 |
| 2 | 0 | 1 | 0 | 1 | 1 | 0 |
| 3 | 0 | 0 | 1 | 0 | 0 | 0 |
| 4 | 0 | 0 | 1 | 0 | 0 | 1 |
| 5 | 0 | 0 | 0 | 0 | 1 | 0 |
【Wiener指数】
n個の原子で構成されている分子について、各原子からある原子までの距離をn×nの行列で示します。
from rdkit import Chem
mol = Chem.MolFromSmiles('C=CC(Cl)CC')
print(Chem.GetDistanceMatrix(mol))
"""
[[0. 1. 2. 3. 3. 4.]
[1. 0. 1. 2. 2. 3.]
[2. 1. 0. 1. 1. 2.]
[3. 2. 1. 0. 2. 3.]
[3. 2. 1. 2. 0. 1.]
[4. 3. 2. 3. 1. 0.]]
"""
これらを行または列方向に足し合わせてtotalが計算され、さらにこれらを足し合わせて1/2をします。これがWiener指数となります。Wiener指数はアルカン類の沸点や密度などの物理特性と相関があることが確認されています (Wiener指数のwikipedia)。
| 0 | 1 | 2 | 3 | 4 | 5 | total | |
|---|---|---|---|---|---|---|---|
| 0 | 0.0 | 1.0 | 2.0 | 3.0 | 3.0 | 4.0 | 13.0 |
| 1 | 1.0 | 0.0 | 1.0 | 2.0 | 2.0 | 3.0 | 9.0 |
| 2 | 2.0 | 1.0 | 0.0 | 1.0 | 1.0 | 2.0 | 7.0 |
| 3 | 3.0 | 2.0 | 1.0 | 0.0 | 2.0 | 3.0 | 11.0 |
| 4 | 3.0 | 2.0 | 1.0 | 2.0 | 0.0 | 1.0 | 9.0 |
| 5 | 4.0 | 3.0 | 2.0 | 3.0 | 1.0 | 0.0 | 13.0 |
W = \frac{1}{2} (13+9+11+9+13) = \frac{1}{2} (55) = 27.5
【Zagreb指数】
rdkitの関数にはなかったようなので、自作しました。Zagreb指数はM1とM2の2つの項があります。
- M1:各原子が結合している原子数の二乗和
- M2:各結合の原子に結合している原子数の積和
今回は上図のClがブランチした化合物とClが直鎖の場合で、値を比較しました。組成式が同じでも、ブランチしている化合物は値が大きくなる結果となりました。この値を用いて、沸点の物性予測ができるそうです (M. Randić, J. Am. Chem. Soc., 1975, 97, 6609.)。そこでは、M2のルートをとった逆数 (Randic指標)を指標とすることで、沸点との直線性を見出しています。ブランチすれば、分子間引力が低下し、沸点が下がる、Randic指標が小さくなると考察しています。2次元の位置関係を数値化したものが、物理特性と相関を持つことは非常に面白い点です。
def rtn_Zagreb_M1(mol):
M1 = 0
for atom in mol.GetAtoms():
bond = atom.GetBonds()
num_bonded = len(bond)
M1 = M1 + num_bonded**2
return M1
def rtn_Zagreb_M2(mol):
M2 = 0
for bond in mol.GetBonds():
atom_begin_idx = bond.GetBeginAtomIdx()
atom_end_idx = bond.GetEndAtomIdx()
atom_begin = mol.GetAtomWithIdx(atom_begin_idx)
atom_end = mol.GetAtomWithIdx(atom_end_idx)
bond_begin = atom_begin.GetBonds()
bond_end = atom_end.GetBonds()
M2 = M2 + len(bond_begin) * len(bond_end)
return M2
mol = Chem.MolFromSmiles('C=CCCCCl')
mol = Chem.MolFromSmiles('C=CC(Cl)CC')
rtn_Zagreb_M1(mol) # 18
rtn_Zagreb_M1(mol) # 20
rtn_Zagreb_M2(mol) # 18
rtn_Zagreb_M2(mol) # 20
3次元
ほとんどの記述子が有料の文献であり、読むことができませんでした。アブストラクトから読み取れた情報をまとめます。
- 幾何学的記述子 (Geometrical descriptor):3D-Wiener index、3D-Balaban index、3D-Harary indexなどが記述子として上げられます。記述子一覧。
- 3D-MoRSE記述子:電子回折に基づく構造の 3D 分子表現 (MoRSE) 記述子は、電子回折 (理論的な散乱曲線の作成に使用) と同じ変換を使用して、3D 座標から 3D 情報を提供します (文献)。記述子一覧。
- WHIM (Weighted Holistic Invariant Molecular)記述子:(x,y,z)-化学物質の分子構造の原子座標から記述子を得ます。記述子一覧。
- GETAWAY (GEometry, Topology, and Atom-Weights AssemblY)記述子:選択された配座における分子の空間座標から計算されるMolecular Influence Matrix (MIM)に基づく記述子です。最初は分子影響マトリックスを算出し、次に幾何学的原子距離と組み合わせることで算出されます。記述子一覧。
立体的な情報を扱うには分子配座が最適化された情報が必要ですが、量子化学計算はrdkitでは行えません。psi4というライブラリならpythonicに量子化学計算が実行できます。量子化学計算で得られた情報を分子記述子として機械学習した報告もあります。
4次元
創薬の分野で発展し、低分子と標的タンパク質との相互作用を予測するために開発されました。立体的な情報 (3次元)に加えて、相互作用を加味することから、4次元と冠されていると思われます。
- GRID:既知の構造の分子上のエネルギー的に有利な結合部位を決定するための計算手順であり、分子間力場 (Molecular Interaction Fields (MIFs))としても知られています。これは主に創薬の分野で利用され、ADMET予測 (Absorption、Distribution、Excretion、Toxicity)や代謝部位予測などに応用されています。
- Volsurf:GRID によって生成された 3D Molecular Interaction Fields (MIF) からの 128 の分子記述子のことです。
- CoMFA(Comparative Molecular Field Analysis) 比較分子場解析:タンパク質と低分子の相互作用を見積もるために開発された回帰分析手法です。
これらの計算は専用のソフトウェアを用いて行われており、rdkitでは計算できません。
【分子記述子の作成のヒント】
ここまで読んでくださった方は、自分で考えた分子記述子を作りたいと思われたかもしれません。rdkitでは、原子の情報を持つAtomオブジェクトと結合の情報を持つBondオブジェクトが用意されており、これらを組み合わせて独自の分子記述子を作成することが可能です。以下にヒントとなりそうなコードを示します。
from rdkit import Chem
from rdkit.Chem import Descriptors
mol = Chem.MolFromSmiles('CC(=O)Oc1ccccc1C(=O)') # アスピリンのsmilesからMolオブジェクトの生成
# 各原子についての情報取得
for atom in mol.GetAtoms():
print("インデックス:{}".format(atom.GetIdx()))
print("\t元素記号:{}".format(atom.GetSymbol()))
print("\t元素番号:{}".format(atom.GetAtomicNum()))
print("\t元素質量:{}".format(atom.GetMass()))
print("\t混成軌道: {}".format(atom.GetHybridization()))
print("\t環内原子か否か: {}".format(atom.IsInRing()))
print("\t芳香環原子か否か: {}".format(atom.GetIsAromatic()))
print("\t結合リストの取得:{}".format(atom.GetBonds()))
print()
"""
インデックス:0
元素記号:C
元素番号:6
元素質量:12.011
混成軌道: SP3
環内原子か否か: False
芳香環原子か否か: False
結合リストの取得:(<rdkit.Chem.rdchem.Bond object at 0x00000227E04F3F30>,)
...
"""
# 各結合の情報取得
for bond in mol.GetBonds():
print("結合のインデックス:{}".format(bond.GetIdx()))
print("\t結合のタイプ:{}".format(bond.GetBondType()))
print("\t芳香環の結合か否か:{}".format(bond.GetIsAromatic()))
print("\t環内結合か否か:{}".format(bond.IsInRing()))
print("\t始点原子の取得:{}".format(bond.GetBeginAtom()))
print("\t始点原子をインデックスで取得:{}".format(bond.GetBeginAtomIdx()))
print("\t終点原子の取得:{}".format(bond.GetEndAtom()))
print("\t終点原子をインデックスで取得:{}".format(bond.GetEndAtomIdx()))
print()
"""
結合のインデックス:0
結合のタイプ:SINGLE
芳香環の結合か否か:False
環内結合か否か:False
始点原子の取得:<rdkit.Chem.rdchem.Atom object at 0x00000227E04F3710>
始点原子をインデックスで取得:0
終点原子の取得:<rdkit.Chem.rdchem.Atom object at 0x00000227E04F3710>
終点原子をインデックスで取得:1
...
"""
# 水素アクセプターと水素ドナーの数
Descriptors.NumHAcceptors(mol) # 3
Descriptors.NumHDonors(mol) # 0
どの分子記述子が良いのか?
これまで紹介した分子記述子はほんの一部です。では数ある分子記述子からどのように選択すればよいのでしょうか?
この文献によれば、目的にあった分子記述子を選択すべきとあります。具体的には、低分子を取り扱う場合はSMILESを利用することが可能ですが、高分子には適しませんので、代わりにBigSMILESが使用できます。不斉反応の場合では光学異性体をそれぞれ異なる表現をする分子記述子が必要ですし、入力した分子と似た特性を持つ分子を出力したいプロジェクトでは分子記述子から元の分子にデコード (分子→分子記述子をエンコード、分子記述子→分子をデコード)できるかが重要です。
目的別に分類してなお、分子記述子を絞り込むことができないかもしれません。その場合はやはり総当りで検討するしかないようです。下図は一般的な最適化方法を示しており、候補となる分子記述子のセットから複数の分子記述子を選択したサブセットを用意し、予測モデルの評価を行います。具体的な方法論としては、evolutionary programming (EP), ant colony optimization (ACO), sequential search (SS), genetic algorithms (GAs)があるようですが、ここでは紹介だけにとどめます。

所感
実際に記事としてアウトプットすることで自分の中でかなり整理することができました。記事を書くまでは、分子記述子=フィンガープリントという考えがあったのですが、今回の調査を通して分子を表現するための手法やそのバリエーションの多さに気付くことができました。また、近年ではDeep Learningを組み合わせた報告もあるようで、そちらも勉強したいと考えています。
最後まで読んでいただき、ありがとうございました。![]()
参考文献
Molecular descriptor (Wikipedia)
RDKitでフィンガープリントを使った分子類似性の判定 (化学の新しいカタチ)
5.3: Chemistry LibreTexts - Molecular descriptor
トポロジカル極性表面積は計算コストの低い推定PSA (化学の新しいカタチ)
RDKitにおける記述子の扱い方をリピンスキーの法則を通して学ぶ (化学の新しいカタチ)
アトムペアやドナーアクセプターペアフィンガープリントは分子の形状を捉えた化学表現 (化学の新しいカタチ)
アトムペアやドナーアクセプターペアフィンガープリントは分子の形状を捉えた化学表現
Molecular Descriptors - BigChem
6.1: Molecular Descriptors - Chemistry LibreTexts
分子記述子計算ソフトウェア mordred の開発 - J-Stage
mordred