概要
深層学習によってある有機物における構造式の二次元画像を入力とした時、その分子式を求める問題を解いてみたい。これは構造式画像に含まれる炭素、水素、酸素、窒素などの原子数を数える回帰問題です。

以前取り組んでいた課題の経緯
そもそも自分は最初、化合物の1次元スペクトルからMorganfingerprintを予測するという課題に取り組んでいました。求めていたMorganfingerprintは化合物の特徴を示す1024bitの0,1のbit配列です。fingerprint同士の類似度を求めれば、測定化合物の同定ができるのでは(少なくとも類似の化合物を検索できるのでは)と考えていました。
しかし、結果は上手く行きませんでした。
予測fingerprintと実際のfingerprintの一致をみるtanimoto係数の平均はいろいろモデルを試してみても0.36が最高で予測fingerprintの精度は低いものでした。
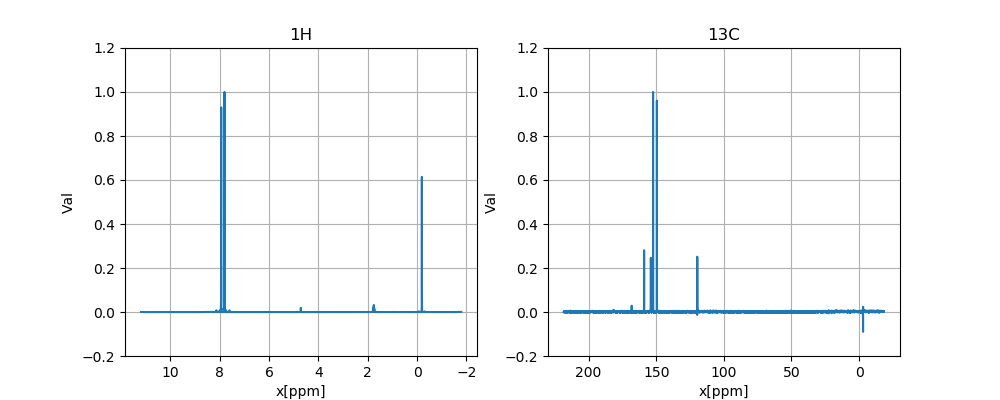
 アデニンの1H,13CのNMRスペクトル
アデニンの1H,13CのNMRスペクトル
これらの原因には以下のような理由が考えられました。
- 学習データが少ない。訓練、テストに用いたデータは800個程度でした。よく深層学習に用いられているのは最低でも数万の単位なのでそれに比べると見劣りします。
- 入力となる測定スペクトルから化合物を同定するには情報量が少ない。そもそも人間であっても1次元スペクトルを解析した段階で(C=)が含まれる、などの特徴量は容易に読み取ることができたとしてもこの1次元スペクトルでは全体構造までは読み取るには情報不足かもしれません。
- fingerprintを予測するのが難しい。fingerprintは1024bitの0,1のbit配列ですが、その持つ情報量はOne-hotベクトルとは比べ物にならないくらい大きくなります。One-hotベクトル1000bitでは$1000=10^3$のラベルを表現しますが、1024bitの0,1のbit配列では$2^{1024}>10^{300}$のラベルを最大で表現できます。実際には分子が実在するために制限があるのでそこまでオーバーではないにせよOne-hotベクトルの予測問題よりかはfingerprintの予測問題を解くのには多くの特徴量(潜在変数)が必要となる可能性は否定できません。
今回の課題
以前の課題に行き詰ってしまったため、もっと簡単な課題に一旦取り組むことにしました。
rdkitを使ってSMILES形式の文字列を構造式画像に変換して、その画像に映った原子の数を数える学習を行ってみました。この課題は以下の特徴により以前の課題の精度が上がらない原因を含まないと推測されます。
- 学習データの入力はPubChemからダウンロードしてきた**30万個(十分な学習データ)**の化合物とします。
- 入力となる構造式の二次元画像は分子構造を理解するのに十分な情報を得られます。
- 出力となる原子を数える作業は分子構造を理解していれば難しい作業ではありません。
手順
0.rdkitのインストール
rdkitをインストールし、構造式にきれいな図を出力するためにPyCairoをインストールします。
1. PubChemからsdfファイルをダウンロードする。
PubChemのsdfファイルは**ここ**からダウンロードできます。もしくはPubChemPyを使うとよいでしょう。
自分の場合はpythonでsdfファイルをCID順にスクレイピングで取得しました。
import urllib.request
for i in range(100):
print('i=%d' % (i))
try:
read_url = 'https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/%d/record/SDF/?record_type=2d&response_type=save&response_basename=Structure2D_CID_%d' % (i,i)
urllib.request.urlretrieve(read_url, './sdf/sdf_%09d.sdf' % (i))
except urllib.error.URLError as e:
pass
2.sdfファイルからSMILESを読み込む
rdkitを使ってsdfファイルを読み込む方法もあります。自分の場合は普通にtxtファイルとして読み込ませました。<PUBCHEM_OPENEYE_CAN_SMILES>の記述がある次の行を読み込み、csvファイルに出力します。
import os.path
f = open('./sdf_data.csv', 'w')
f.write('Number,CAN_SMILES\n')
for i in range(100):
print('i=',i)
fname = './sdf/sdf_%09d.sdf' % (i)
if os.path.isfile( fname ):
f2 = open(fname, encoding='utf-8')
content = f2.readlines()
for line_num, line in enumerate(content):
if '<PUBCHEM_OPENEYE_CAN_SMILES>' in line:
f.write('%d,%s' % (i, content[line_num+1]))
f2.close()
break
f.close()
これによって以下のSMILESデータの抽出を行いました。
Number,CAN_SMILES
1,CC(=O)OC(CC(=O)[O-])C[N+](C)(C)C
2,CC(=O)OC(CC(=O)O)C[N+](C)(C)C
3,C1=CC(C(C(=C1)C(=O)O)O)O
4,CC(CN)O
5,C(C(=O)COP(=O)(O)O)N
6,C1=CC(=C(C=C1[N+](=O)[O-])[N+](=O)[O-])Cl
7,CCN1C=NC2=C1N=CN=C2N
8,CCC(C)(C(C(=O)O)O)O
9,C1(C(C(C(C(C1O)O)OP(=O)(O)O)O)O)O
10,C1C(NC2=C(N1)NC(=NC2=O)N)CN(C=O)C3=CC=C(C=C3)C(=O)NC(CCC(=O)O)C(=O)O
...
3.学習を行う
上記のSMILESデータをrdkitを用いて$(300,300,3)$の大きさの画像に変換し入力とします。また、出力を分子式の['C', 'H', 'N', 'O', 'S', 'P', 'Si', 'Na', 'F', 'Cl', 'Br', 'I']の12種類の原子の個数とします。
modelはXceptionを用い、損失はloss='mean_squared_error'としました。
学習中rdkitがClの2価のSMILES形式データをmol形式に変換できないと警告を出しますが、学習には支障ないので無視します。(このrdkitの警告を消す方法があれば教えてください…)
# CNNを構築(Xception)
from keras.applications.xception import Xception
inputs = Input(shape=(300,300,3)) # data_size (300,300,3)
x = Xception(weights=None,input_shape=(300,300,3), include_top=False, pooling='avg')(inputs)
y = Dense(12, activation='linear')(x)
model = Model(inputs, y)
4.学習後に予測結果を確認する
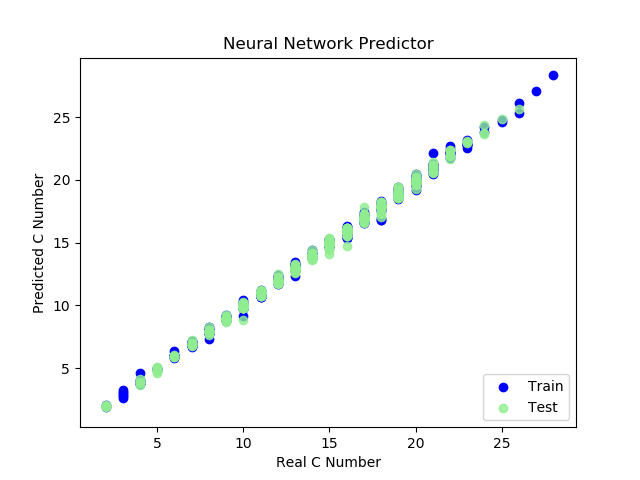
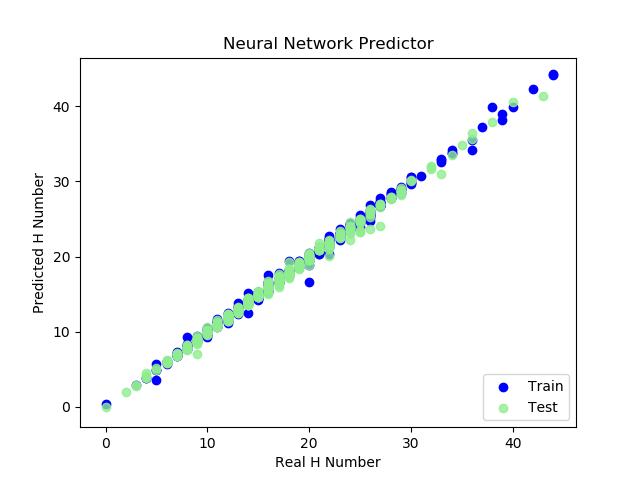
テストデータの予測を出力してみて原子の数え上げができているか確認しました。
結果は以下のとおりです。中でも水素(H)の数え上げは構造式画像中に水素原子が明示的には記載されないため難易度が少し高いのではと思いましたが、水素を含む原子数の予測はほぼ成功していました。
上手く行かなった例ではP(リン)を検出できず、またCH3も一つ検出に失敗しています。
リン酸(PO4)はmol質量が大きく、今回の学習範囲では学習が他より行われていないのかもしれません。
| 化合物の構造式 | 実際の分子式 | 予測の分子式 |
|---|---|---|
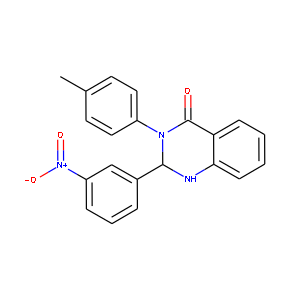 |
C21H17N3O3 | C21.0H16.9N3.0O2.9 |
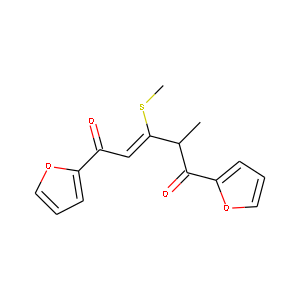 |
C15H14O4S1 | C15.1H14.2O3.8S1.0 |
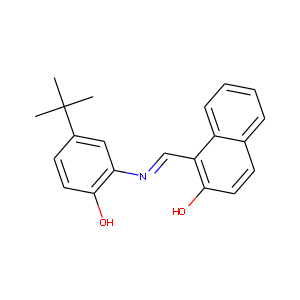 |
C21H21N1O2 | C20.8H21.1N1.0O2.0 |
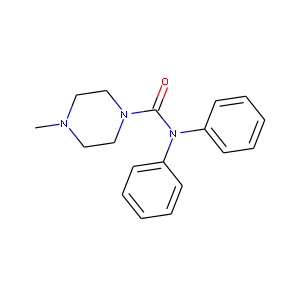 |
C18H21N3O1 | C17.8H20.8N2.9O1.2 |
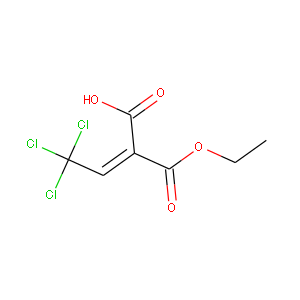 |
C7H7O4Cl3 | C7.3H7.1O4.0Cl2.7 |
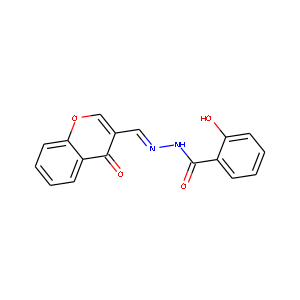 |
C17H12N2O4 | C16.6H11.8N1.9O4.1 |
| 上手く行かなかった例 | ||
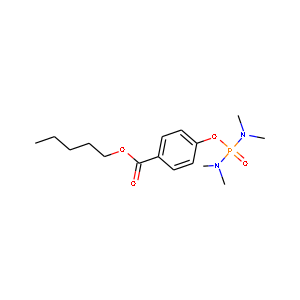 |
C16H27N2O4P1 | C14.7H24.1N1.8O3.7 |
 |
||
 |
まとめ
構造式画像から化学式を導出するという簡単な課題は解くことが可能であることを確認できました。
この結果から前回の行き詰ってしまった課題の改善すべき点を考えていきたいと思います。
学習コード
# coding: cp932
import keras
from keras.layers import Input, Concatenate, GlobalAveragePooling2D
from keras.models import Model
from keras.layers.core import Dense, Dropout, Activation, Flatten
from keras.callbacks import LearningRateScheduler, ModelCheckpoint
from keras.preprocessing.image import img_to_array, array_to_img
import numpy as np
import pandas as pd
from sklearn.model_selection import train_test_split
import matplotlib.pyplot as plt
import os.path
import re
from rdkit import Chem
from rdkit.Chem import Draw, rdDepictor, rdMolDescriptors
max_MW = 400 # MWの最大値
load_weights_path = './weight_param.hdf5' # 重みファイルのパス
# SMILESの読み込み
df = pd.read_csv('sdf_data.csv')
SMILES = df['CAN_SMILES'].values
# arrayに変換
SMILES = np.asarray(SMILES)
SMILES_train, SMILES_test = train_test_split(SMILES, test_size=0.30, random_state=110)
print(SMILES_train.shape, SMILES_test.shape)
# 読み込みデータのメモリ開放
del df, SMILES
class DataGenerator():
def __init__(self, X_input):
self.X_input = X_input
self.atom = ['C', 'H', 'N', 'O', 'S', 'P', 'Si', 'Na', 'F', 'Cl', 'Br', 'I']
self.reset()
def reset(self):
self.X = []
self.Y = []
def cal_img(self, mol):
# 構造式画像を生成
img = Chem.Draw.MolToImage(mol, size=(300, 300))
return img_to_array(img)[:,:,:3]
def chem_count(self, input):
atom = ['C', 'H', 'N', 'O', 'S', 'P', 'Si', 'Na', 'F', 'Cl', 'Br', 'I']
count = {'C': 0, 'H': 0 ,'N': 0, 'O': 0, 'S': 0, 'P': 0, 'Si': 0, 'Na': 0,
'F': 0, 'Cl': 0, 'Br': 0, 'I': 0}
input = input + 'Z' # 末尾に英語大文字を追加
for i in range(len(atom)):
if re.search(atom[i] + '\d{1,3}[A-Z]', input): #原子+数字(1~3桁)+英語大文字を含む場合
iterator = re.finditer(atom[i] + '\d{1,3}[A-Z]', input)
for match in iterator:
count[atom[i]] = int(match.group()[len(atom[i]):-1])
elif re.search(atom[i] + '[A-Z]',input): #原子+英語大文字を含む場合
count[atom[i]] = 1
return count
def make_structural_image(self, batch_size=10):
while True:
for i in range(len(self.X_input)):
# SMILES形式をmol形式に変換
mol = Chem.MolFromSmiles(self.X_input[i], sanitize=True)
if not mol is None: # molファイルがNoneなら弾く
# 二次元位置を計算
rdDepictor.Compute2DCoords(mol)
# mol質量を計算
MW = Chem.rdMolDescriptors._CalcMolWt(mol)
# mol形式から分子式を計算
chem_Formula = Chem.rdMolDescriptors.CalcMolFormula(mol)
# 分子式から原子数をカウント
count = self.chem_count(chem_Formula)
# X,Yを追加
if MW < max_MW and count['C'] > 1.0:
# 炭素原子が2個以上含まれるmol質量400以下の化合物であること。
(self.X).append(self.cal_img(mol))
(self.Y).append([count[self.atom[i]] for i in range(len(self.atom))])
# X,Yの値を返す
if len(self.X) == batch_size:
inputs = np.asarray(self.X)
outputs = np.asarray(self.Y)
self.reset()
yield inputs, outputs
# DataGeneratorを生成
train_datagen = DataGenerator(SMILES_train)
test_datagen = DataGenerator(SMILES_test)
batchsize = 16
# CNNを構築(Xception)
from keras.applications.xception import Xception
inputs = Input(shape=(300,300,3)) # data_size (300,300,3)
x = Xception(weights=None,input_shape=(300,300,3), include_top=False, pooling='avg')(inputs)
y = Dense(12, activation='linear')(x)
model = Model(inputs, y)
# モデル表示
model.summary()
# コンパイル
model.compile(loss='mean_squared_error',
optimizer='Adam',
metrics=['accuracy'])
# 保存した重みを読み込む場合
if os.path.isfile(load_weights_path):
model.load_weights(load_weights_path)
# コールバック
callbacks = []
# 学習率の低減
base_lr = 1e-3
lr_decay_rate = 1 / 3
lr_steps = 4
epochs = 200
callbacks.append(keras.callbacks.LearningRateScheduler(lambda ep:
float(base_lr * lr_decay_rate ** (ep * lr_steps // epochs))))
# モデル出力
fpath = './weights_{epoch:03d}-{loss:.2f}-{val_loss:.2f}.hdf5'
callbacks.append(ModelCheckpoint(filepath = fpath, monitor='val_loss',
verbose=0, save_best_only=True, mode='auto'))
# 学習
history = model.fit_generator(
generator=train_datagen.make_structural_image(batch_size=batchsize),
steps_per_epoch=int(len(SMILES_train) / batchsize / 100),
epochs=epochs,
verbose=1,
validation_data=test_datagen.make_structural_image(batch_size=batchsize),
validation_steps=100,
callbacks=callbacks)
# モデルと重みの保存
model.save_weights('weight_param.hdf5')
model.save("model.h5")
# 学習グラフのプロット
plt.figure()
plt.plot(history.history['loss'])
plt.plot(history.history['val_loss'])
plt.title('model loss')
plt.xlabel('epoch')
plt.ylabel('loss')
plt.legend(['loss', 'val_loss'], loc='upper right')
plt.ylim([0,0.5])
plt.show()
#########################
# 予測
# 500データの予測計算
X_train, Y_train = next(train_datagen.make_structural_image(batch_size=500))
X_test, Y_test = next(test_datagen.make_structural_image(batch_size=500))
Y_predict_train = model.predict(X_train)
Y_predict_test = model.predict(X_test)
# 予測のプロット
atom = ['C', 'H', 'N', 'O', 'S', 'P', 'F', 'Cl']
for i in range(len(atom)):
plt.figure()
plt.scatter(Y_train[:,i], Y_predict_train[:,i], label = 'Train', c = 'blue')
plt.title('Neural Network Predictor')
plt.xlabel('Real ' + atom[i] + ' Number')
plt.ylabel('Predicted ' + atom[i] + ' Number')
plt.scatter(Y_test[:,i], Y_predict_test[:,i], c = 'lightgreen', label = 'Test', alpha = 0.8)
plt.legend(loc = 4)
plt.show()
# 50件のテスト結果を出力
atom = ['C', 'H', 'N', 'O', 'S', 'P', 'Si', 'Na', 'F', 'Cl', 'Br', 'I']
for i in range(50):
img = array_to_img(X_test[i])
real = ''
pred = ''
for j in range(len(atom)):
if round(Y_test[i,j]) > 0:
real += atom[j] + str(round(Y_test[i,j]))
if round(Y_predict_test[i,j],1) > 0.5:
pred += atom[j] + str(round(Y_predict_test[i,j],1))
img.save('test_result%04d_%s_%s.png' % (i, real, pred))