はじめに
HTSLIbとは、SAM/BAM/CRAM/VCF/BCF などを操作するC言語のライブラリです。
Samtools や Bcftools など多くのツールでライブラリとして利用されています。
この記事では、pileup の構造体と関数を見ていきます。
本記事では、インターネット上に日本語の情報が少ない HTSlib の内容を、AI支援を用いて解析・検証しながらまとめました。内容には誤りがある可能性があります。お気づきの点があれば、コメント欄でご指摘ください。
Pileup とはなにか
「Pileup」は「山積み」を意味し、リードを座標ごとに積み上げて観察する手法を指します。Pileup とは「山積み」を意味する 名詞 であり、積むという動作ではなく、仕事や書類が積み上がった状態であることに気をつけてください
IGVの画面では多数のリードが積み重なっていると思います。これが pileup です。
HTSlibでは pileup 操作に関連する一連の構造体と関数を提供しています。
どんなときに使うの
特定の座標のリードの情報を集計したいときに使います。
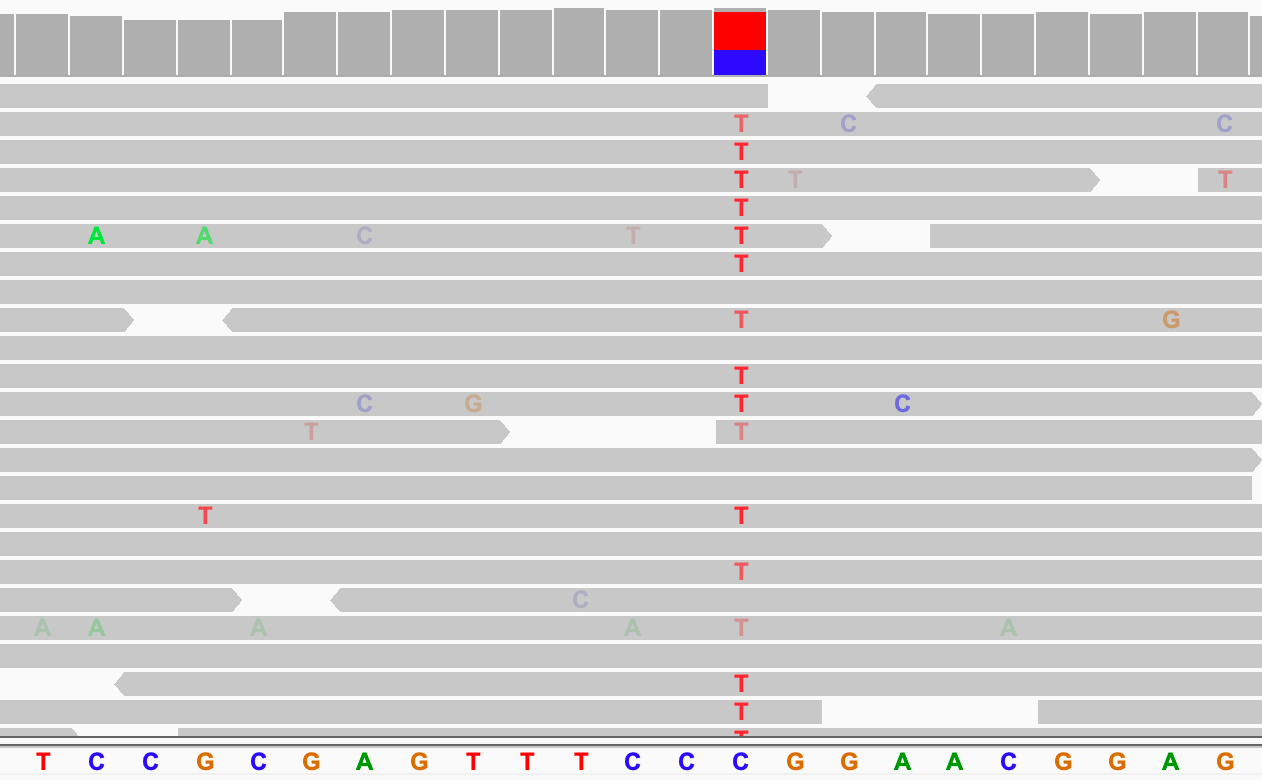
出典:https://igv.org/doc/desktop/UserGuide/img/alignments-mismatches.png
上の図では、リファレンスの塩基がCで、Tのリードが混ざっている箇所があります。
このように特定のゲノム座標(一塩基単位)に着目してデータを集計したいとき、pileup が有用です。たとえば以下のようなプログラムで pileup が使われます。
- 変異コーラー - 各ゲノム座標における塩基を走査し、SNPやInDelを抽出する。
- 塩基修飾の集計 - 各ゲノム座標ごとに塩基修飾を集計する。
- カバレッジ解析 - 各ゲノム座標ごとにリード深度を算出する。
Pileup が向いていないケース
詳細な塩基情報(≒CIGAR解析)を必要としないケースでは pileup はオーバースペックです。高速に近似値を求めたい場合は、pileupを使わずに自分でリードを一本ずつ処理したほうがよいケースも多いと思います。
たとえば、mosdepth のファストモードではCIGAR情報を展開せず、リードの始点と終点のみを利用してカバレッジを高速に算出しています。
sam.h における定義箇所
HTSlibは扱う内容(ゲノム情報)の難しさ・実装の複雑さ(多くは高速化のための最適化)の2つの面で、なかなか手ごわいライブラリですが、ヘッダーファイルにドキュメントがしっかり書かれている関数が多いのです。けれども、Pileupの項目では2025年11月現在、あまりドキュメントが書かれていません。(後述するように、現在は利用されていないフィールドがあったり、塩基修飾の用に最近整備が進められたフィールドがあることを考えると、仕様が完全に固定されていない可能性があります。)
スニペット:sam.h の PileupとMpileupのセクション
そこで、pileupの理解と利用は諦めていたのですが、最近はAIがライブラリを丁寧に説明してくれるようになりました。本記事はAIに聞いた内容を元に情報を整理しています。重要な構造体と関数は以下の通りです。
- 構造体
-
bam1BAMレコードを表す構造体 -
bam_pileup1_t特定のゲノム座標においてBAM1(ポインタ)、リードにおける位置、インデル、塩基修飾などの情報を持つ構造体
-
| 関数 | 概要 | 主な用途 |
|---|---|---|
bam_plp_init |
Pileupイテレータを初期化 | コールバックの登録 |
bam_plp_push |
BAMレコードを追加 | 手動でレコード供給 |
bam_plp_next |
次の座標のpileupを取得 | 結果の取り出し |
bam_plp_auto |
pushとnextを自動処理 | コールバック利用時 |
これらの構造体と関数の詳しい内容は後述します。
SAM・BAM 形式のおさらい
SAM・BAM はアライメントのレコードをまとめたデータ形式です。SAMはタブ区切りテキスト形式で、BAMはBGZFで圧縮されたバイナリ形式です。
BAMファイルを染色体と座標の順序にソートすると、インデックスファイル(.bai)を作成できるようになります。
インデックスファイルは「ゲノム座標の位置」と「BGZF圧縮ブロックの位置(オフセット)」の対応を持ちます。これにより圧縮ファイルでありながら、指定された領域に効率的にランダムアクセスが可能になります。
関心領域のレコードを抽出する
HTSlibでは、iter_query 系の関数を使うことで、関心のある範囲のレコード(リード)を取り出して処理することができます。
#include <stdio.h>
#include <htslib/sam.h>
int main(int argc, char *argv[])
{
// 0. 宣言
const char *bamfile = argv[1];
samFile *fp = sam_open(bamfile, "r");
bam_hdr_t *hdr = sam_hdr_read(fp);
hts_idx_t *idx = sam_index_load(fp, bamfile);
// 1. 指定領域をイテレータで取得
hts_itr_t *itr = sam_itr_querys(idx, hdr, "chr1:100000-200000");
// 2. BAM レコードを読み込む
bam1_t *b = bam_init1();
while (sam_itr_next(fp, itr, b) >= 0) {
printf("%s\t%s\t%d\n",
bam_get_qname(b),
hdr->target_name[b->core.tid],
b->core.pos + 1);
}
// 3. 後始末
bam_destroy1(b);
hts_itr_destroy(itr);
hts_idx_destroy(idx);
bam_hdr_destroy(hdr);
sam_close(fp);
}
Pileup を使わずに縦に見る場合
Pileupを使わずに、特定の座標で塩基を集計する場合を考えて見ましょう。
sam_itr_next を使えば関心領域のリードを、座標の順番に取り出すことは可能です。
しかし、SAM・BAMのレコードの位置 pos は、あくまでアラインメントの開始位置です。(参照配列上のアライメント左端。内部実装は 0-based、概念的には 1-based)
そこで、リードの塩基が、参照配列のどこに対応しているかは、一本ずつ自分でCIGAR解析を行って確認する必要があります。実際には指定した座標の塩基が、挿入であったり欠失であるケースもあり面倒です。
別の座標で集計を行いたい場合、sam_itr_hext を繰り返し呼び出すことになり、効率的ではない可能性があります。
Pileupの使い方
手動で bam_plp_push / bam_plp_next する方法
構造体の内容を把握してから使い方を理解したい方も多いと思います。その場合は本項は飛ばして次項を先に読んでください。
Pileupを使ってみましょう。
まずは初期化 bam_plp_initでコールバックを行わないケースをAIに書いてもらいました。
#include <stdio.h>
#include <htslib/sam.h>
int main(int argc, char *argv[])
{
// 0. 宣言
const char *bamfile = argv[1];
samFile *fp = sam_open(bamfile, "r");
bam_hdr_t *hdr = sam_hdr_read(fp);
hts_idx_t *idx = sam_index_load(fp, bamfile);
// 1. 指定領域をイテレータで取得
hts_itr_t *itr = sam_itr_querys(idx, hdr, "chr1:100000-101000");
// 2. pileup イテレータを初期化
bam_plp_t plp = bam_plp_init(NULL, NULL); // コールバック不要
// 3. BAM レコードを読み込んで pileup に供給
bam1_t *b = bam_init1();
while (sam_itr_next(fp, itr, b) >= 0) {
bam_plp_push(plp, b);
}
// 4. pileup を取り出して出力
const bam_pileup1_t *p;
int tid, pos, n;
while ((p = bam_plp_next(plp, &tid, &pos, &n)) != NULL) {
// tid = 染色体ID, pos = 0-based
printf("%s\t%d\t%d\n", hdr->target_name[tid], pos + 1, n);
}
// 5. 後始末
bam_destroy1(b);
bam_plp_destroy(plp);
hts_itr_destroy(itr);
hts_idx_destroy(idx);
bam_hdr_destroy(hdr);
sam_close(fp);
}
まず、bam_plp_t plp = bam_plp_init(NULL, NULL); としてイテレータを初期化します。
次に、while (sam_itr_next(fp, itr, b) >= 0) { bam_plp_push(plp, b); } として bam_plp_push を行います。
最後に while ((p = bam_plp_next(plp, &tid, &pos, &n)) != NULL) { } を行います。
初期化された pileup の入力ストリームに、ユーザーが自分で BAMレコード を供給している箇所に注目してください。HTSlibでは、Pileup解析をする場合、単にBAMファイルを開くだけでは不十分であり、bam_plp_push 関数を使って、自分でBAMレコードを追加する必要があります。(この記事の冒頭でpileupが動詞ではなく名詞であることを強調したのはそのためです)。これによって、Pileup解析に使うアライメントレコードを自分で選べる柔軟性がありますが、このままでは使いにくいので、通常は bam_plp_t を初期化する際に、自動でレコードを追加してくれるコールバック関数を登録します。
このようにコールバック関数(および後述の遅延評価)を含む複雑なAPIを持つ関数群を、他言語からバインディングで利用すると大変煩雑になり、またコールバック関数はPythonやRubyなど他言語で実装することになりますので一般的にはパフォーマンスも悪化します。そこで、多くの言語のTHSLibバインディングでは、Pileupには未対応であるか、あらかじめ事前に静的言語で記述されたコールバック関数を用いるなど、大幅な簡略化が行われることが多いです。
たとえば、Pythonではコールバック関数はCython(cpythonではない)であらかじめ3種類(all, nofilter, samtools)定義されており、stepper引数で指定します。
for column in bam.pileup("chr1", 100, 200, stepper="samtools"):
pass
-
all-BAM_FUNMAP、BAM_FSECONDARY、BAM_FQCFAIL、BAM_FDUPのフラグが設定されているリードをスキップします -
nofilter- すべてのリードを使用し、フィルタリングを行いません -
samtools- samtools pileupコマンドと同じフィルタリングとリード処理を行います
デフォルトでは samtools が指定されます。これは暗黙のうちに以下の条件でフィルタリングを行うので少し注意が必要です。これは知らないと間違えそうですね。
- フラグフィルタ:
BAM_FUNMAP|BAM_FSECONDARY|BAM_FQCFAIL|BAM_FDUP - 最小ベースクオリティ:
13 - オーバーラップを無視:
True- read1とread2が重なっている場合に、重複カウントを避ける
- より高いベースクオリティを持つ方のベースのみをカウント
- オーファンリードを無視:
True- ペアエンドリードで片方だけがマッピングされている場合(オーファンリード)、そのマッピングされた側も無視する
結果(つまりPileup(名詞))は bam_plp_next で取得します。
戻り値は専用の構造体ではなく、bam_pileup1_t の配列(上のサンプルコードだと *p)として表現されます。bam_plp_next 関数は、bam_plp_push で提供されるBAMレコードの位置より手前の、確定した位置について結果(Pileup)を返すことができます。結果を返却できないときは NULL を返します。
bam_plp_auto を使う方法
次に、初期化でコールバック関数を登録し、bam_plp_auto を使う場合のコードを示します。
bam_plp_auto は bam_plp_push と bam_plp_next を内部で自動的に呼び出す高レベルAPIです。
#include <stdio.h>
#include <htslib/sam.h>
// コールバック用のデータ構造
typedef struct {
samFile *fp;
hts_itr_t *itr;
} plp_data;
// コールバック関数: イテレータから次のレコードを読み込む
static int read_bam(void *data, bam1_t *b) {
plp_data *d = (plp_data *)data;
return sam_itr_next(d->fp, d->itr, b);
}
int main(int argc, char *argv[])
{
// 0. 宣言
const char *bamfile = argv[1];
samFile *fp = sam_open(bamfile, "r");
bam_hdr_t *hdr = sam_hdr_read(fp);
hts_idx_t *idx = sam_index_load(fp, bamfile);
// 1. 指定領域をイテレータで取得
hts_itr_t *itr = sam_itr_querys(idx, hdr, "chr1:100000-101000");
// 2. コールバック用データを準備
plp_data data = { .fp = fp, .itr = itr };
// 3. pileup イテレータをコールバック付きで初期化
bam_plp_t plp = bam_plp_init(read_bam, &data);
// 4. auto を使って pileup を取り出して出力
const bam_pileup1_t *p;
int tid, pos, n;
while ((p = bam_plp_auto(plp, &tid, &pos, &n)) != NULL) {
// tid = 染色体ID, pos = 0-based
printf("%s\t%d\t%d\n", hdr->target_name[tid], pos + 1, n);
}
// 5. 後始末
bam_plp_destroy(plp);
hts_itr_destroy(itr);
hts_idx_destroy(idx);
bam_hdr_destroy(hdr);
sam_close(fp);
}
bam_plp_auto は bam_plp_push と bam_plp_next の実行を自動化します。
内部で bam_plp_push と bam_plp_next を交互に呼び出すことでメモリを効率を良く使うことができます。bam_plp_next は bam_pileup_1 を返却出来ない場合は、NULLを返却しますが、この挙動はほとんどコストがかかりません。bam_plp_next を呼び出すと同時にレコードのCIGAR解析が行われます。ここでは遅延評価を行うことで計算効率を改善しているのですが(bam_plp_push側ではなくbam_plp_next側で処理を行う)、C言語で書かれているため、見慣れないAPIが難しく感じられます。
Pileupの構造体
Pileupの使い方を見ましたので、構造体をもう少し詳しく見てみましょう。
Pileupには2種類の構造体があります。
bam_pileup1_t
/*! @typedef
@abstract pileup位置をカバーする1つのアライメントを表す構造体
@field b アライメントへのポインタ
@field qpos pileup位置でのリードベースの位置(0ベース)
@field indel indelの長さ; indelなしの場合は0、挿入の場合は正、欠失の場合は負
@field level 「ビューア」モードでのリードのレベル
@field is_del パディングされたリード上のベースが欠失の場合は1
@field is_head クエリシーケンスの最初のベースの場合は1
@field is_tail クエリシーケンスの最後のベースの場合は1
@field is_refskip パディングされたリード上のベースがCIGAR N操作の一部の場合は1
@field aux (bcf_call_gap_prep()で使用される)
@field cigar_ind 処理されたばかりのCIGAR操作のインデックス
@discussion bam_plbuf_push()とbam_lplbuf_push()も参照してください。
この2つの関数の違いは、前者はbam_pileup1_t::levelを設定しないのに対し、
後者は設定する点です。Levelはアライメントビューアの実装に役立ちますが、
これを計算するにはいくらかのオーバーヘッドがあります。
*/
typedef struct bam_pileup1_t {
bam1_t *b;
int32_t qpos;
int indel, level;
uint32_t is_del:1, is_head:1, is_tail:1, is_refskip:1, /* reserved */ :1, aux:27;
bam_pileup_cd cd; // generic per-struct data, owned by caller.
int cigar_ind;
} bam_pileup1_t;
bam_pileup_1 は bam_1 構造体をラッピングして必要な情報を追加したものと考えられます。各フィールドについてコメントします。
b は bam_1 へのポインタです。qpos はおそらく query position の略で、リード側からみた塩基の位置(0-based)です。indel は直後の位置に、挿入・欠失がある場合にその長さを表します(後述の is_del との違いに注意)。level フィールドはゲノムブラウザの表示に使われていた値で、現在の htslib では設定されません。samtools の tview では、このlevelが利用されていることから、以前の実装の名残だと考えられます。さらに、is_del, is_head, is_tail, is_refskip などのビットフィールド(int型などを分割してフラグに使用するもの)が続きます。is_del は現在の位置が欠失である場合に1になります。is_head や is_tail はアラインメントにおけるheadやtailではなくquery配列のheadやtailであることを示します。is_refskip は、CIGAR配列でNのケースを示しますが、多くはRNA-Seqにおいてエクソン間のイントロンを意味します。aux は bcftools で使われており互換性のために残されていますが、htslibにはフィールドを設定する関数はありません。bam_pileup_cd は後述のようにさまざまなデータをもたせられる共用体ですが、修飾塩基としての使い方が今後拡大すると思われます。 cigar_ind はCIGARのインデックスです。
bam_pileup_cd
typedef union {
void *p;
int64_t i;
double f;
} bam_pileup_cd;
修飾塩基情報やユーザー定義の一時データなどを保持するための汎用共用体です。bam_pileup1_t の Aux とでもいうべきものです。拡張性のために int64 や double も格納できるようになっています。しかし多くのケースでは void *p が使われていると思います。
特に、に修飾塩基情報を格納して利用するケースが多いです。
bam_plp_constructor() コンストラクタで、 bam_parse_basemod を呼び出して、修飾塩基情報をパースするコールバック関数を登録すると、bam_plp_push した際に、修飾塩基を解析してくれるようになります。
修飾塩基をパースする例
以下に DeepWikiに書いてもらったサンプルコードを提示します。
#include <stdio.h>
#include <htslib/sam.h>
// コールバック用のデータ構造
typedef struct {
samFile *fp;
sam_hdr_t *h;
} plp_dat;
// BAMレコードを読み込むコールバック関数
static int readaln(void *data, bam1_t *b) {
plp_dat *dat = (plp_dat *)data;
return sam_read1(dat->fp, dat->h, b);
}
// コンストラクタ: 修飾塩基の状態を初期化
int pileup_cd_create(void *data, const bam1_t *b, bam_pileup_cd *cd) {
hts_base_mod_state *m = hts_base_mod_state_alloc();
if (bam_parse_basemod(b, m) < 0) {
hts_base_mod_state_free(m);
return -1;
}
cd->p = m; // bam_pileup_cdに状態を格納
return 0;
}
// デストラクタ: 修飾塩基の状態を解放
int pileup_cd_destroy(void *data, const bam1_t *b, bam_pileup_cd *cd) {
hts_base_mod_state_free(cd->p);
return 0;
}
int main(int argc, char **argv) {
samFile *in = sam_open(argc > 1 ? argv[1] : "-", "r");
sam_hdr_t *h = sam_hdr_read(in);
// pileupイテレータの初期化
plp_dat dat = { .fp = in, .h = h };
bam_plp_t iter = bam_plp_init(readaln, &dat);
// コンストラクタ/デストラクタを登録
bam_plp_constructor(iter, pileup_cd_create);
bam_plp_destructor(iter, pileup_cd_destroy);
const bam_pileup1_t *p;
int tid, pos, n;
// pileupを走査
while ((p = bam_plp_auto(iter, &tid, &pos, &n)) != 0) {
printf("%s\t%d\t", sam_hdr_tid2name(h, tid), pos);
for (int i = 0; i < n; i++, p++) {
if (p->is_del) {
putchar('*');
continue;
}
// 塩基を出力
uint8_t *seq = bam_get_seq(p->b);
unsigned char c = seq_nt16_str[bam_seqi(seq, p->qpos)];
putchar(c);
// 修飾塩基を取得
hts_base_mod_state *m = p->cd.p;
hts_base_mod mod[5];
int nm = bam_mods_at_qpos(p->b, p->qpos, m, mod, 5);
if (nm > 0) {
putchar('[');
for (int j = 0; j < nm && j < 5; j++) {
if (mod[j].modified_base < 0)
// ChEBI番号の場合
printf("%c(%d)%d", "+-"[mod[j].strand],
-mod[j].modified_base, mod[j].qual);
else
// 1文字コードの場合
printf("%c%c%d", "+-"[mod[j].strand],
mod[j].modified_base, mod[j].qual);
}
putchar(']');
}
}
putchar('\n');
}
bam_plp_destroy(iter);
sam_close(in);
sam_hdr_destroy(h);
return 0;
}
まとめ
本記事では HTSlib における pileup API の構造と基本的な使い方を整理しました。
特に bam_plp_init, bam_plp_push, bam_plp_next の関係性と使い方、修飾塩基を扱う bam_pileup_cd の利用方法を紹介しました。
- 複数のファイルを扱うMpileup
- さまざまなプログラミング言語のhtslibバインディングにおける、pileup対応状況の比較
については、今後余裕があれば、またまとめたいと思います。