Biopyton からClustalW2を使って系統樹を作成する方法について自分用にメモ.
といっても内容はほとんどBiopython Tutorial and Cookbookに書かれていることを日本語に訳しただけ.
ClustalW2 をインストール
まず,ClustalW2 をダウンロードする.Macの場合には.dmgをマウントして得られるbinファイルを/bin下に置く.
系統樹を作成したい株を用意する
次にClustalW2を使用する株のデータを用意する.今回はメタリジウム(Metarhizium)の系統樹をRibosome biogenesis protein YTM1をもとに作成する.ファイルはUniProtからダウンロードした.
使用した株は次の通り.
- Metarhizium acridum (strain CQMa 102)
- Metarhizium anisopliae BRIP 53293
- Metarhizium guizhouense ARSEF 977
- Metarhizium album ARSEF 1941
- Metarhizium robertsii
- Metarhizium rileyi RCEF 4871
Basketに追加した後,FASTA形式でダウンロードする.今回はuniprot-yourlist.fastaという名前で保存した.
BiopythonでClustalW2を実行する
用意した株のデータに対してBiopythonからClustalW2をかける.
from Bio.Align.Applications import ClustalwCommandline
clustalw_cline = ClustalwCommandline("clustalw2", infile="uniprot-yourlist.fasta")
stdout, stderr = clustalw_cline()
するとuniprot-yourlist.alnとuniprot-yourlist.dndの2つのファイルが生成されている.そこでBiopythonのPhyloモジュールを使ってdndファイルの方を読み込み,系統樹を描く.
from Bio import Phylo
tree = Phylo.read("uniprot-yourlist.dnd", "newick")
Phylo.draw(tree)
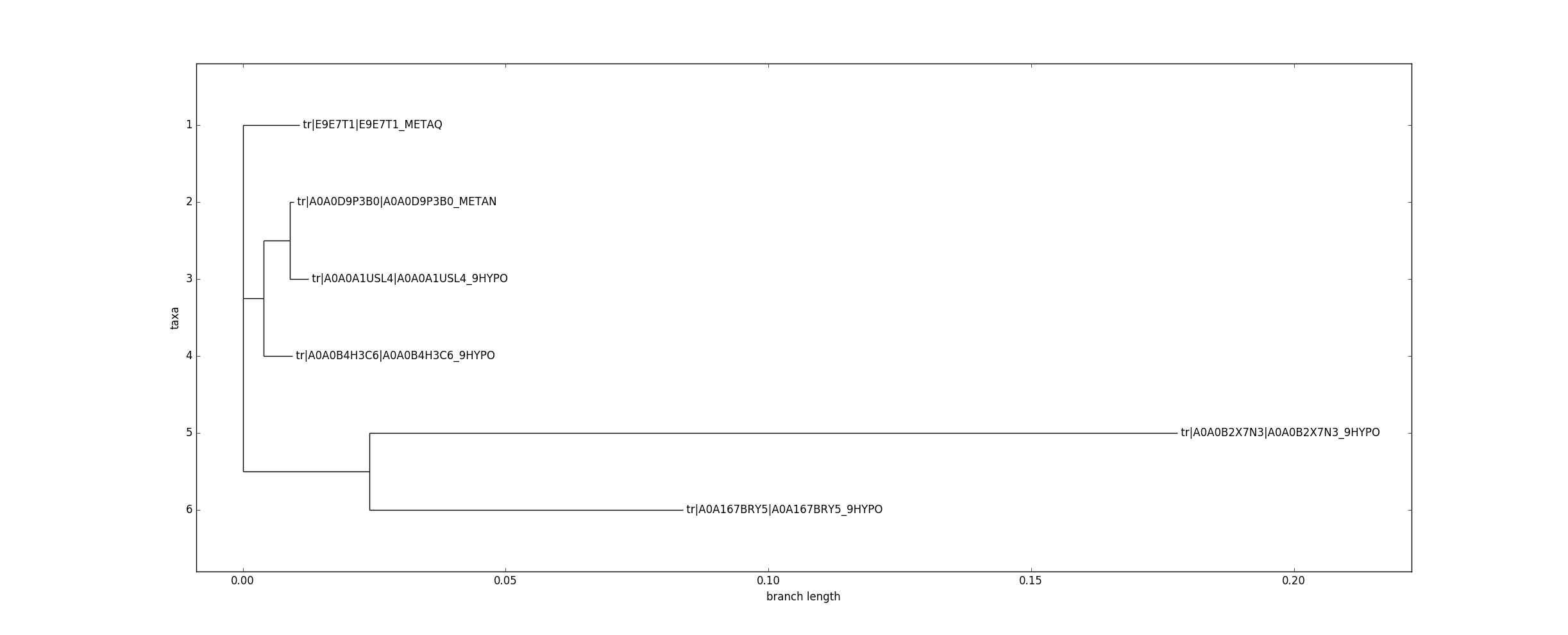
draw関数の代わりにdraw_ascii関数を使うと系統樹をアスキーアートで出力してくれる.
_ tr|E9E7T1|E9E7T1_METAQ
|
| , tr|A0A0D9P3B0|A0A0D9P3B0_METAN
|,|
_||| tr|A0A0A1USL4|A0A0A1USL4_9HYPO
||
|| tr|A0A0B4H3C6|A0A0B4H3C6_9HYPO
|
| ______________________________________ tr|A0A0B2X7N3|A0A0B2X7N3_9HYPO
|_____|
|______________ tr|A0A167BRY5|A0A167BRY5_9HYPO
Exited with code=0 in 1.1
参考文献
Peter J. A. Cock, Tiago Antao, Jeffrey T. Chang, Brad A. Chapman, Cymon J. Cox, Andrew Dalke, Iddo Friedberg, Thomas Hamelryck, Frank Kauff, Bartek Wilczynski, Michiel J. L. de Hoon: “Biopython: freely available Python tools for computational molecular biology and bioinformatics”. Bioinformatics 25 (11), 1422–1423 (2009). doi:10.1093/bioinformatics/btp163,
Eric Talevich, Brandon M. Invergo, Peter J.A. Cock, Brad A. Chapman: “Bio.Phylo: A unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython”. BMC Bioinformatics 13: 209 (2012). doi:10.1186/1471-2105-13-209