本記事は2019年秋に書かれたものになります。最新版とは仕様が異なる可能性があります。
はじめに
本稿は岐阜大学3年生向けの講義「ゲノム生物学」で説明される16S rRNAアンプリコンシーケンスを実行するための補助教材です。学部4年生が書いています。免責事項として、すべて正しいことが書けているとは考えておりませんので、実習向けとして捉えてください。ここでは、菌叢の棒グラフを作ることをゴールにします。希薄化、多様性解析などの工程を飛ばし、最低限の理解を行うことが講義の目的です。研究を行うための解析については当方別の記事、Qiime2のチュートリアル、DRY解析本を参照ください。
Qiime2公式サイト: https://qiime2.org/
メッセージ
間違いを指摘してくださるプロの皆さま、メールかメッセージください。ツイッターが最速で返信可能です。もしくはコマンドを動かしても、うまくできない場合はご連絡ください。
Twitter @danryo_official
動作条件
- Macbook Air Early 2015, macOS Mojave 10.14.6
- Miniconda conda 4.7.12
- Excelが入っていること
早速やってみる
今回はQiime2をインストールして、軽く菌叢解析をしていこうと思います。Qiime2をMacで落とす際はminicondaの利用がベスト。本稿ではQiime2の公式説明を読みながら、分かりにくいところを追加して書いている感じです。したがって、本稿で不明な点があればhttps://docs.qiime2.org/2019.10/ を見てください。
動作環境として、macOSにminicondaを入れているものとします。入れ方はググると無限に出てきます。PCが苦手な後輩も一人でできたので、飛ばします。Latestバージョン推奨なので、新しく入れた人は最新版を入れていると知っていますが、念の為アップデートします。あと、wgetコマンドが使えない人はhomebrewかcondaで入れておいてください。
$ conda update conda
$ conda install wget
どのディレクトリでもいいと思います。当方はホームディレクトリに落としました。
$ wget https://data.qiime2.org/distro/core/qiime2-2019.10-py36-osx-conda.yml
$ conda env create -n qiime2-2019.10 --file qiime2-2019.10-py36-osx-conda.yml
ymlファイルが残ってウザいと思う人は消してください。
$ rm qiime2-2019.10-py36-osx-conda.yml
qiime2-2019.10のインストールには少し時間がかかるようです。たくさんのファイルがPCに入れられていきます。
Collecting package metadata (repodata.json): done
Solving environment: done
Downloading and Extracting Packages
bioconductor-shortre | 5.2 MB | ##################################### | 100%
bleach-3.1.0 | 110 KB | ##################################### | 100%
jpeg-9c | 237 KB | ##################################### | 100%
terminado-0.8.2 | 23 KB | ##################################### | 100%
bioconductor-biocgen | 695 KB | ##################################### | 100%
scipy-1.3.1 | 16.1 MB | ##################################### | 100%
scikit-bio-0.5.5 | 1.2 MB | ##################################### | 100%
matplotlib-3.1.0 | 6 KB | ##################################### | 100%
harfbuzz-1.9.0 | 769 KB | ##################################### | 100%
pyzmq-18.1.0 | 464 KB | ##################################### | 100%
r-gtable-0.3.0 | 420 KB | ##################################### | 100%
r-munsell-0.5.0 | 244 KB | ##################################### | 100%
gneiss-0.4.6 | 41 KB | ##################################### | 100%
zeromq-4.3.2 | 571 KB | ##################################### | 100%
r-formatr-1.7 | 165 KB | ##################################### | 100%
emperor-1.0.0b20 | 557 KB | ##################################### | 100%
測ってないので分かりませんが、ダウンロードに結構かかります (軽く10分以上)。ここはPCとネット環境依存です。
終わると
#
# To activate this environment, use
#
# $ conda activate qiime2-2019.10
#
# To deactivate an active environment, use
#
# $ conda deactivate
と出てきます。指示に従い、
$ conda activate qiime2-2019.10
をします。使うたびにactivateするようです。baseがqiime2-2019.10に変わります。うまくできていれば、help画面が出てきます。
$ qiime --help
これでインストール完了です。
Qiime2のチュートリアル
Moving Pictures Tutorialを参考にコマンドを書いてきます。一応設定としてSingle end (SE) でシーケンスされたデータということです。Pair endのチュートリアルも別にあります。いつになるか分かりませんが、書いてみる予定です。
多分このWikiを読む人は「バリバリ実験マンだから、NGSの解析なんてやったことねえよ」な人か、「学部生 (もしくは修士マン) で研究室で勉強しろって言われた」な人だと思います。したがって、どうやって16S ampliconが用意されているのかはご存知だと思います。今回のデータはEMPに記載されている方法が用いられています。EMPと呼ばれるシーケンシングスタンダードです。僕なんかよりもずっと詳しいかと思いますので、調製に関しては省略します。Caporaso et al. (2011)のデータということで、ざっくりヒトの腸内細菌をHiSeqで読んでいるようです。
まとめると、データとして用いるのはEMP法で調製されたSEリードです。ヒトの腸内細菌のデータです。
データのダウンロード
データのダウンロードをします。Qiime2に必要なデータは3種類です。
データの中身について記載したTSVファイル、シーケンス結果を含んだFASTQファイル、シーケンスのうちバーコード配列のみを含んだFASTQファイルです。通常MiSeqやHiSeqを利用する場合、1ランで複数のサンプルを解析します。したがって、バーコード配列を利用して振り分けます。TSVファイルの中身を利用することで、後々条件区ごとにデータを分けて作図できます。データをダウンロードしてから、TSVファイルをExcelで開いて実際に見てみるといいと思います。
チュートリアルデータを作る前に作業用ディレクトリを作成しておきましょう。当方は見やすいようにDesktopに作成しました。作業用ディレクトリ内に配列だけを入れるemp-single-end-sequencesディレクトリも作成します。
$ cd Desktop/
$ mkdir qiime2-moving-pictures-tutorial
$ cd qiime2-moving-pictures-tutorial
$ mkdir emp-single-end-sequences
続いて、データを落とします。落とし方は好きなようにやるといいと思います。URLをクリックしてDownloadディレクトリに落とすのもありですし、wgetコマンドを使うのもありだと思います。
# sample_metadata.tsvをダウンロード
$ wget https://data.qiime2.org/2019.10/tutorials/moving-pictures/sample_metadata.tsv
# barcodes.fastq.gzとsequences.fastq.gzをemp-single-end-sequences内にダウンロード
$ cd emp-single-end-sequences/
$ wget https://data.qiime2.org/2019.10/tutorials/moving-pictures/emp-single-end-sequences/barcodes.fastq.gz
$ wget https://data.qiime2.org/2019.10/tutorials/moving-pictures/emp-single-end-sequences/sequences.fastq.gz
同じようにやっていれば、qiime2-moving-pictures-tutorialディレクトリの中にTSVファイルとemp-single-end-sequencesディレクトリがあり、emp-single-end-sequencesディレクトリ内にfastq.qzファイルが2つある状態です。
ファイルをインポート
Qiime2ではfastqファイルというファイル形式ではなく、qzaファイルという特殊な形式に変更して作業を行なっていきます。この際、インポートという作業を行います。作業ディレクトリはqiime2-moving-pictures-tutorialにしてください。
$ cd qiime2-moving-pictures-tutorial/
$ qiime tools import \
--type EMPSingleEndSequences \
--input-path emp-single-end-sequences \
--output-path emp-single-end-sequences.qza
さて、それぞれ検討していくと
--typeではデータタイプを選択します。EMPSingleEndSequencesというタイプを選ぶとSequence.fastq.qzとBarcodes.fastq.qzというファイルが--input-pathであるemp-single-end-sequencesにある場合importできるようです。
# --typeで入れられるデータタイプ
portable-types
DeblurStats
DistanceMatrix
EMPPairedEndSequences
EMPSingleEndSequences
ErrorCorrectionDetails
FeatureData[AlignedSequence]
FeatureData[Differential]
FeatureData[Importance]
FeatureData[PairedEndSequence]
FeatureData[Sequence]
FeatureData[Taxonomy]
FeatureTable[Balance]
FeatureTable[Composition]
FeatureTable[Frequency]
FeatureTable[PercentileNormalized]
FeatureTable[PresenceAbsence]
FeatureTable[RelativeFrequency]
Hierarchy
MultiplexedPairedEndBarcodeInSequence
MultiplexedSingleEndBarcodeInSequence
PCoAResults
Phylogeny[Rooted]
Phylogeny[Unrooted]
Placements
QualityFilterStats
RawSequences
SampleData[AlphaDiversity]
SampleData[BooleanSeries]
SampleData[ClassifierPredictions]
SampleData[DADA2Stats]
SampleData[FirstDifferences]
SampleData[JoinedSequencesWithQuality]
SampleData[PairedEndSequencesWithQuality]
SampleData[Probabilities]
SampleData[RegressorPredictions]
SampleData[SequencesWithQuality]
SampleData[Sequences]
SampleEstimator[Classifier]
SampleEstimator[Regressor]
SeppReferenceDatabase
TaxonomicClassifier
UchimeStats
めちゃめちゃ多いので、使うやつだけ抜粋すると
# EMPPairedEndSequences
マルチプレックスのforward.fastq.qz, reverse.fastq.qz, barcodes.fastq.qz
# 'SampleData[SequencesWithQuality]'
他のソフトでdemultiplexed済みのfastq.gzファイル (同時に複数読み込める)
# 'SampleData[PairedEndSequencesWithQuality]'
他のソフトでdemultiplexed済みのfastq.gzファイル (Forward, Reverse) (ペアになる名前R1, R2がついていれば同時に複数読み込める)
Demultiplex
先に述べた通り、NGSは通常複数のサンプルを1回のランで解析します。なので、1つのデータとして出てきますが、中には複数の条件のデータが入っています。これをバーコード配列を使って分けていきます。弊研究室ではタグ分けと呼んでおりますが、増やしたrRNA領域配列の外側に配列を仕込んでおき、これをもとに分けます。したがって、ここでのバーコード配列の意味は16S rRNA配列のことではないです。通常キットを使う場合、キットに配列が仕込まれていますのであまり気にする事ないのですが、きちんと入っています。なお、キットのバーコード配列よりもさらにバーコード配列を仕込む場合、sabreによってさらに分ける必要があります。弊研究室では自作スクリプトで分けていますが、そのうちsabreも紹介する予定です。特に受託で解析する場合でなおかつ配列数が十分すぎる場合、この方法で1ランの費用で5~6ランなどできるので安く済みます。詳しくは「DNA情報で生態系を読み解く」という本に書いてあります。
早速Demultiplexしてみます。
$ qiime demux emp-single \
--i-seqs emp-single-end-sequences.qza \
--m-barcodes-file sample_metadata.tsv \
--m-barcodes-column barcode-sequence \
--o-per-sample-sequences demux.qza \
--o-error-correction-details demux-details.qza
qzaファイルは中身が見れないファイルになります。そこで、これをqzvに書き換えるという方法がQiimeでファイルの中身を見るために必要だそうです。といっても簡単です。以下のコマンドを打つだけです。
$ qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
書き換わったQZVファイルはGoogle Chromeなどを使うと簡単に確認できます。以下のコマンドでビジュアル化してみます。
qiime tools view demux.qzv
僕の場合はhtmlファイルをGoogle Chromeで開く設定にしているので、このコマンドを使うとChromeが立ち上がります。
どうやらhtmlファイルは処理中に書き出されており、それを開けという命令のようです。ファイルは以下のような感じになります。
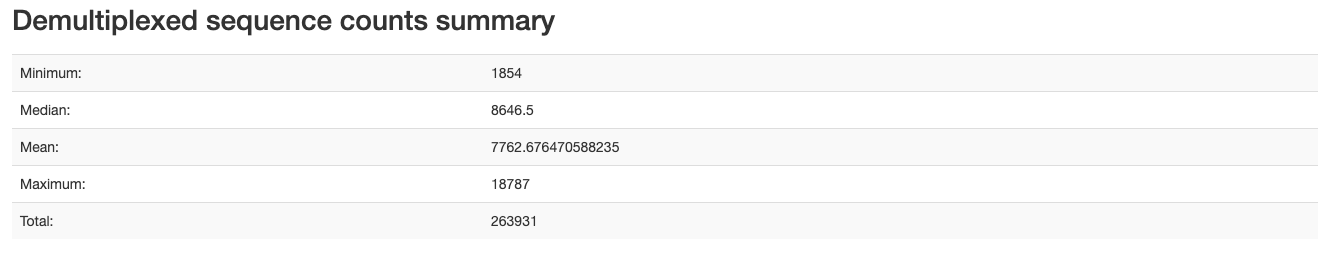
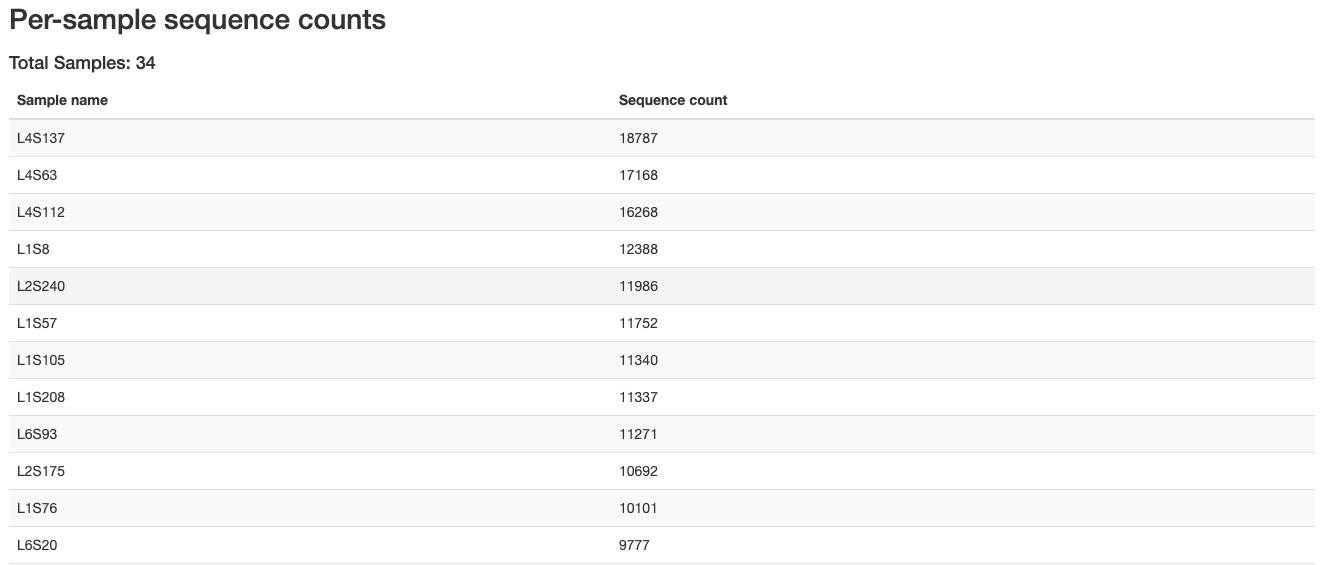
各条件毎のリード数などが得られます。PDF形式でも書き出すことができるようです。
Quality ControlとFeature Tableの作製
Qiime2のクオリティーコントロールでは3つの方法があり、それぞれ選ぶことができます。それがDADA2、Deblur、basic quality-score-based filteringです。なお、私はよく分かりません。DADA2の論文を見ましたが、よく分からない鳥居みたいな記号が出てきました。それぞれ論文があるので、気になる人は読んでください。今回は最も有名なDADA2でやってみたいと思います。
DADA2 Denoise
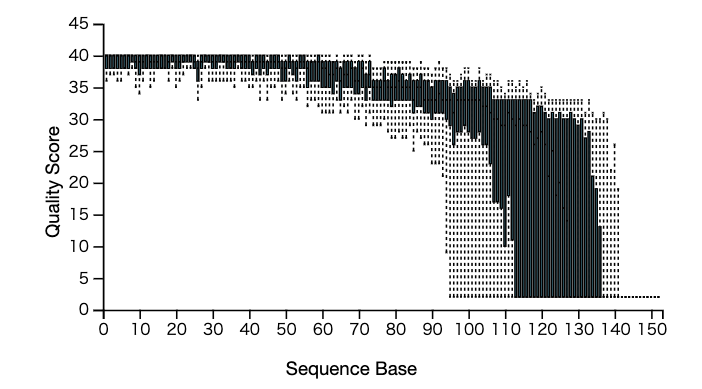
最初にトリミングします。先に示した以下の図はリードをランダムに1万個抽出し各リードの位置毎にどの程度のクオリティーがあるかを示した図です。

これを見ると、120くらいでかなり低くなっていることが明白です。対して、リードの最初の方はクオリティーがかなり高いです。そこで、truncateといって、不要な配列部分を切り落とします。--p-trim-leftで5'末端側から何塩基を切り落とすか、--p-trunc-lenでスタートから何塩基目は配列を切り落とすかを決めます。チュートリアルだと120 bpで--p-trunc-lenすることになっていますが、僕は80でやりたいと思います。先輩によると、ここで選択した配列より短いものは全て捨てられてしまうようなので、16Sでは問題ないですがITSなどに汎用させる際は注意が必要です。また、qzaをzipに置き換えて解凍すると中身が確認できます。ここからの作業で中身 (ファイルそのもの) が見たい人はやってみてください。
$ qiime dada2 denoise-single \
--i-demultiplexed-seqs demux.qza \
--p-trim-left 0 \
--p-trunc-len 80 \
--o-representative-sequences rep-seqs-dada2.qza \
--o-table table-dada2.qza \
--o-denoising-stats stats-dada2.qza
この作業は10分くらいかかるそうです。出てきたデータを確認してみます。
$ qiime metadata tabulate \
--m-input-file stats-dada2.qza \
--o-visualization stats-dada2.qzv
$ qiime tools view stats-dada2.qzv
出てきたデータは以下の通りでした。
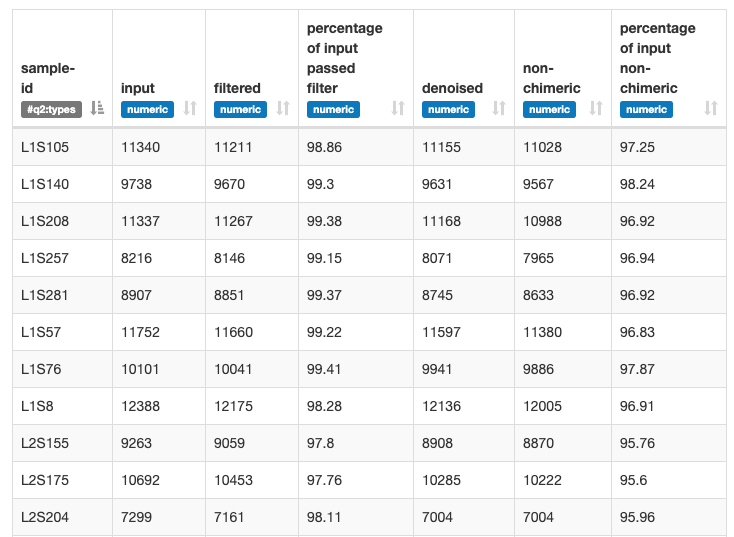
このプロセスでやってくれていることとして、PhiXと呼ばれるMiSeqやHiSeqで必要なコントロールの除去、先に指定した条件でのトリミング、キメラ配列の除去、さらにクラスタリングまで済ませています。このクラスタリング情報を使って、Feature Tableと呼ばれるTaxonomy解析における最重要情報を作成します。チュートリアルでは、ここで名前を変更していました。不必要ですが、コマンドをそのままコピペで打っている人がいれば、この作業をしてください。
$ mv rep-seqs-dada2.qza rep-seqs.qza
$ mv table-dada2.qza table.qza
Feature Tableを作ります。Feature Tableには2種類の情報があります。一つはFrequencyを述べたもの、もう一つが配列を述べたものです。データ量を小さくするため、配列が100%一致の場合は配列を消去して1つにするようです。Frequencyを述べるFeature Tableが必要なのはこのためで、例えば配列Aが10回登場しましたというようなデータだということです。ここではfeature-table summarizeとfeature-table tabulate-seqsを使って、how many sequences are associated with each sample and with each feature, histograms of those distributionsという情報が含まれるtable.qzvとa mapping of feature IDs to sequences, and provide links to easily BLAST each sequence against the NCBI nt databaseという情報が含まれるrep-seqs.qzvを作ります。
$ qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.tsv
$ qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
データを見るのが早いと思うので、早速ビジュアライズしてみます。まずはtable.qzvから。ここではsample-metadata.tsvを利用しますので、sample-metadata.tsvにつけられたindex毎にデータをまとめて解析できます。例えば、male, femaleという条件を入れておけば、その条件に従ってデータを分けてみられるということです。OTUでの特徴なようです。
$ qiime tools view table.qzv
続いて、rep-seqs.qzvを見てみます。こちらはFASTAでダウンロード可能な代表配列 (?) が得られるようです。そのままダイレクトにBLASTもできる仕様になっています。
$ qiime tools view rep-seqs.qzv
よく見る棒グラフを作る
ここまでの情報で足りないのはTaxomomyの情報です。つまり、データベースが足りません。
そこで、Qiime2が配布しているものを利用します。
$ wget https://data.qiime2.org/2019.10/common/gg-13-8-99-515-806-nb-classifier.qza
これを使って、先の情報と照らし合わせて、棒グラフを作成します。
まずは棒グラフにする前まで持っていきます。
$ qiime feature-classifier classify-sklearn \
--i-classifier gg-13-8-99-515-806-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
$ qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
これをビジュアライズします。データとしては各配列にID情報が載せられ、それに対して相同性検索を行い、どの程度一致したかを見ているようです。
$ qiime tools view taxonomy.qzv
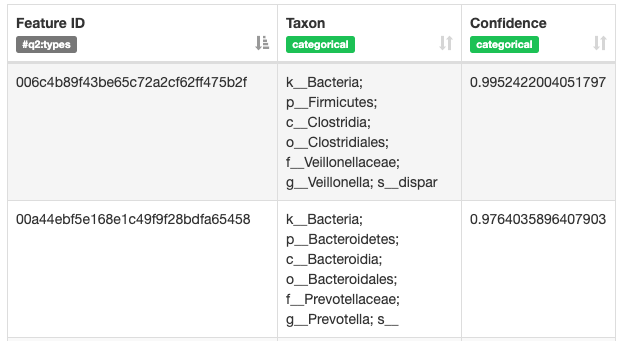
あとはこの情報にFrequencyを合わせて、1つのOTUにどの程度含まれているかを計算することで最終的に細菌量 (割合) が計算されます。実際にやってみます。
$ qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization taxa-bar-plots.qzv
これをビジュアライズします。これでいつもよく見るバープロットの完成です。お疲れ様でした。
$ qiime tools view taxa-bar-plots.qzv
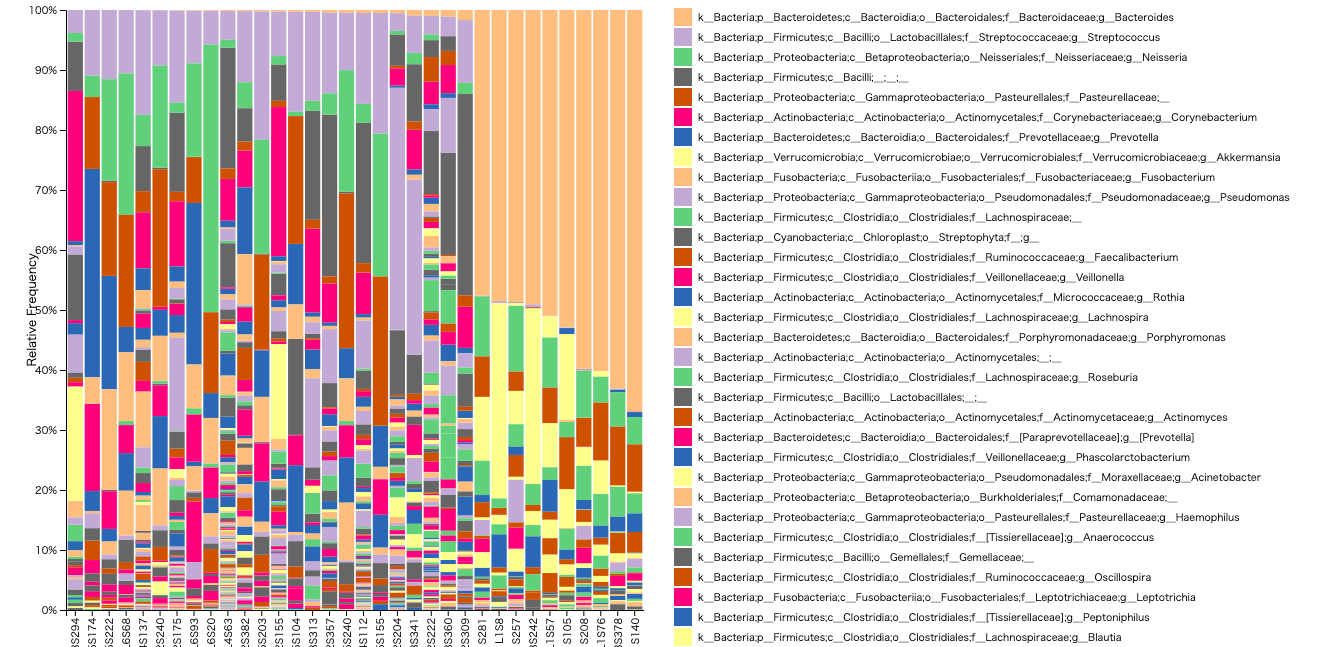
終わりに
本稿で紹介した以外のことをQiime2で解析することは可能です。ここに載っていましたが、以下のようなデータを出力することが可能なようです。まだまだ勉強すべきことは多そうです。
- demux.qzv - シーケンスとクオリティの概要
- table.qzv - フィーチャー(OTU)
- rep-seqs.qzv - 代表配列
- jaccard_emperor.qzv - Jaccard 指数(類似度距離)
- bray_curtis_emperor.qzv - Bray-Curtis 指数(類似度距離)
- unweighted_unifrac_emperor.qzv - Unweighted UniFrac (質的距離)
- weighted_unifrac_emperor.qzv - Weighted UniFrac (量的距離)
- faith-pd-group-significance.qzv - 試料グループ間の系統学的多様性
- evenness-group-significance.qzv - 試料グループ間の均等度
- unweighted-unifrac-body-site-significance.qzv - 試料グループ間の類似度検定
- taxonomy.qzv - フィーチャーの系統分類
- taxa-bar-plots.qzv - 試料の系統構成比率