導入
創薬、機能性材料、農薬、化粧品、フレーバー…
「分子」を扱う産業は多岐にわたりますが、共通しているのは
- 候補となる分子の数が途方もなく多いこと
- すべてを実験することは到底できないこと
です。
ケモインフォマティクス(Cheminformatics/化学情報学) は、
化学構造や反応の情報をデジタルデータとして扱い、
物性予測や分子設計・スクリーニングをデータ駆動で支援する分野
です。
本記事では、コードや数式には踏み込まずに、
- ケモインフォマティクスとは何か
- どんなデータ・タスク・ワークフローがあるのか
- 現在どのように使われているのか
- 課題と今後の方向性
を、化学専攻でないエンジニアや学生にもイメージしやすいように整理します。
TL;DR
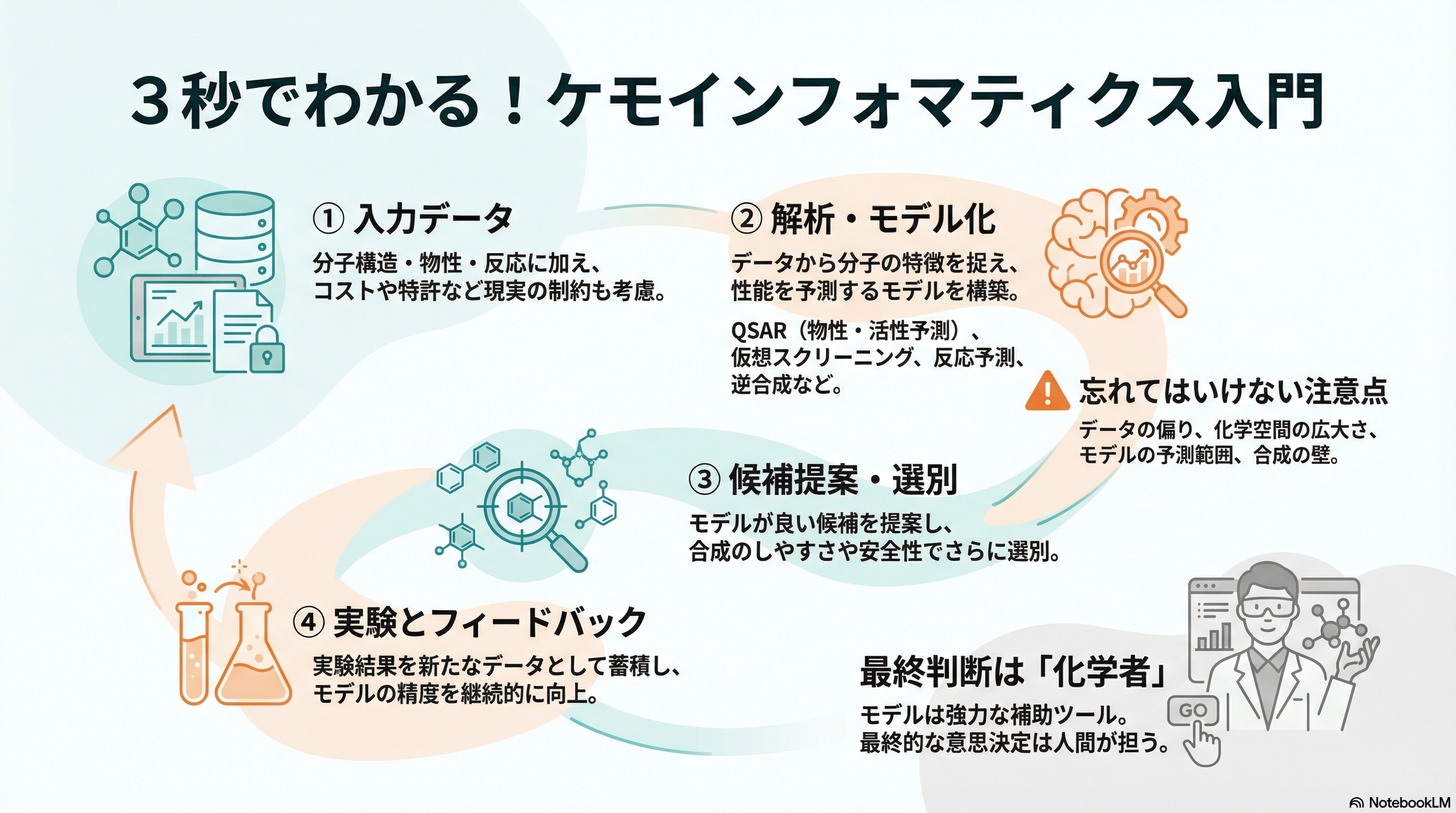
- ケモインフォマティクスは、化学構造や反応データを情報として扱い、分子の性質予測や設計を支援する分野。
- 主な対象は、低分子化合物の構造、物性データ、反応経路、活性や毒性の情報など。
- 代表的なタスクは、**物性・活性予測(QSAR)、仮想スクリーニング、反応予測、逆合成(レトロシンセシス)**など。
- 創薬や材料設計で、実験前に候補を絞り込むための道具として広く使われている。
- 課題は、データの質・バイアス・化学空間の広大さ・合成可能性の考慮など。
- 今後は、深層学習や生成モデル、実験自動化との連携による「設計〜合成〜評価のループ高速化」が重要になっていく。
ケモインフォマティクスとは?
一言でいうと
ケモインフォマティクスは、
分子構造や化学反応をコンピュータで扱いやすい形に表現し、
物性や活性の予測・分子設計・反応経路探索を支援する技術・研究分野
です。
主な目的は、実験や試作の前に
- 「どの分子が狙った性質を持ちそうか」
- 「どの構造が効きやすく、どの構造が危なそうか」
- 「この分子を作るための合成ルートはありそうか」
といった問いに対して、「当たり」をつけることです。
対象とする分子・領域
ケモインフォマティクスの主な対象は、
- 低分子化合物(医薬品候補、農薬、香料など)
- 高分子やオリゴマー(ポリマー材料など)
- 化学反応や合成ルート
などです。
創薬のイメージが強いかもしれませんが、機能性材料や電子材料、界面活性剤など、幅広い分野で利用されています。
どんなデータを扱うのか
ケモインフォマティクスで扱うデータは、大きく次のように分けられます。
1. 分子構造データ
- 原子の種類と数
- 原子同士の結合関係(どの原子がどの原子とどう結合しているか)
- 場合によっては 3 次元構造(立体配置)
計算機上では、
- SMILES などの文字列表現
- MOL/SDF などの構造ファイル
- グラフ(ノード=原子、エッジ=結合)
といった形で表現されます。
2. 物性・活性データ
- 物理化学的性質(溶解度、融点、分配係数など)
- 生物学的活性(ターゲットに対する作用の強さ)
- 安全性(毒性、発がん性など)
- ADME 関連(吸収、分布、代謝、排泄に関わる特性)
分子構造とセットで記録され、「この構造はこういう性質を持つ」というペアが多数集まります。
3. 反応・合成データ
- どの化合物とどの化合物を反応させると、何が生成するか
- 反応条件(溶媒、触媒、温度、時間など)
- 収率や副反応などの情報
これらは、反応予測や逆合成ルート探索に使われます。
4. その他の関連情報
- コストや入手性(原料が簡単に手に入るか)
- 特許情報(どの構造が既に特許でカバーされているか)
- 規制・法令(有害物質指定など)
これらは、設計段階で「現実的かどうか」を判断する上で重要です。
代表的なタスクとユースケース
1. 物性・活性予測(QSAR)
QSAR(Quantitative Structure–Activity/Property Relationship) は、
分子構造(入力)から、物性値や活性値(出力)を予測する
取り組みです。
- 構造から数値的特徴量(記述子)を計算する
- 過去データをもとに回帰・分類モデルを作る
- 新しい分子の特徴量を入れて、性質を予測する
という流れで、
- 「毒性が少なそうな構造」
- 「溶けやすい/溶けにくい構造」
- 「標的タンパク質に効きそうな構造」
などを絞り込むのに使われます。
2. 仮想スクリーニング
巨大な化合物ライブラリ(現実に合成済みのもの・仮想的なもの)に対して、
- QSAR モデルや類似度検索を使って、条件を満たす候補を探す
- 必要ならドッキング計算なども併用して、ターゲットとの結合しやすさを評価する
というプロセスが 仮想スクリーニング です。
実験でのスクリーニングは時間もコストもかかるため、先にコンピュータ上で候補を絞り込み、有望そうなものだけを実験に回すのが一般的です。
3. 反応予測と逆合成(レトロシンセシス)
ケモインフォマティクスは、反応の世界にも広がっています。
-「この 2 つの分子を反応させたら、何ができそうか?」
-「この目的分子を作るには、どんな原料からどんなステップで作ると良さそうか?」
といった問いに対して、
- 既知の反応データベースやルールに基づく方法
- 機械学習による条件・生成物の予測
などを用いて答えを提案するのが、反応予測・レトロシンセシス(逆合成)です。
4. ライブラリ設計・多様性解析
- 化合物ライブラリ全体の分布や多様性を解析する
- 特定の化学空間(構造の範囲)のカバレッジを見る
- 似たような構造ばかりになっていないかチェックする
これにより、「効率良く新しい構造空間を探索できているか」を判断し、ライブラリを設計・改良することができます。
典型的なワークフロー
ケモインフォマティクスのプロジェクトは、大まかには次のような流れで進みます。
1. データ収集とクリーニング
- 社内の実験データ、公開データベース、文献などからデータを集める
- 構造の重複や誤登録をチェックし、標準化する
- 明らかにおかしい値や矛盾のあるレコードを確認する
化学データは、単位や表記ゆれ、塩/フリー体の扱いなど、細かなルールが多く、ここがまず大変なところです。
2. 分子表現と特徴量設計
- 分子をグラフや文字列として表現する
- 元素種・結合種・部分構造などから、記述子を計算する
- 必要に応じて、3D 構造に基づく特徴量も導入する
近年は、グラフニューラルネットワークなど、分子グラフから直接特徴を学習する手法も広く使われています。
3. モデル構築と評価
- 回帰・分類モデルで物性・活性を予測する
- 交差検証や外部テストセットで、汎化性能を確認する
- 化学的に妥当な範囲で外挿が効くかどうかもチェックする
ここで重要なのは、「数字上の精度」だけでなく、
- どの領域では当たりやすく、どの領域では外しやすいか
- どの構造に対してはモデルが自信を持てるか
といった性質も理解することです。
4. 分子設計・スクリーニングへの応用
- 設計したい条件を満たす分子を、既存ライブラリから探す
- 生成モデルや最適化手法を使って、新しい構造候補を提案する
- 合成可能性や安全性、コストなども加味して、実験候補を選ぶ
この段階で、人間の化学者の知見とモデルの提案を組み合わせて、「現実的に試すべき候補」を議論していきます。
現状の活用状況と課題
創薬・材料での実用
- 製薬企業やケミカル企業では、ケモインフォマティクスは既に日常的な道具になっている
- ヒット化合物の探索や、物性の荒い目安取り、類似構造探索などで活用されている
- 材料分野でも、特定の物性(導電性、耐熱性、界面活性など)に関するモデルが使われつつある
一方で、「モデルが提案した構造がそのまま薬になる」という段階ではなく、
モデル:候補の絞り込みと直感の補助
人間:最終判断と合成戦略の立案
といった役割分担が現実的です。
データの質とバイアス
- 実験条件や測定系が異なるデータを混ぜると、モデルが混乱しやすい
- 活性ありの化合物ばかり載っているデータセットでは、「効かない構造」を十分に学習できない
- 特定の構造クラスに偏ったデータで学習すると、他の領域へはうまく外挿できない
このため、単にデータ量を増やすだけでなく、どの領域をどれだけカバーしているかという観点が重要になります。
化学空間の広大さ
- 理論的には存在しうる分子構造の数は膨大で、実際にデータがあるのはそのごく一部
- モデルは「見たことがあるような構造」に対しては強いが、「まったく新しい構造」には自信を持ちにくい
ここは、マテリアルズインフォマティクスやバイオインフォマティクスとも共通する課題です。
合成可能性・現実性
- モデルが提案する分子が、実際に合成できるとは限らない
- 合成できても、工程数が多すぎたり、危険な原料が必要になる場合もある
そのため、最近は「合成しやすさ」や「反応条件の現実性」を加味したモデル化・評価が重視されています。
今後の方向性
深層学習・生成モデルとの統合
- グラフニューラルネットワークで分子グラフを直接扱う手法
- 分子を連続ベクトル空間に埋め込んで最適化する手法
- 条件付き生成モデルで、特定の物性を満たす分子候補を自動生成する手法
などにより、「表現・予測・生成」がシームレスにつながりつつあります。
実験自動化との連携
- ロボット合成や自動測定装置と組み合わせ、設計〜合成〜評価のループを自動で回す
- モデルが提案した候補を自動で合成・評価し、その結果でモデルを継続的に改善する
こうした「自律的な化学実験」との連携は、今後の大きなテーマの一つです。
安全性・規制・説明可能性
- 毒性や環境影響を含めて、総合的に「リスクの低い分子」を設計するニーズが高まっている
- 規制や法令への対応のため、モデルの挙動を説明し、根拠を示すことが求められる場面も増えている
「当たればよい」から一歩進んで、「なぜそう判断したのか」「責任を持てるか」が問われるフェーズに入りつつあります。
これから学びたい人へのヒント
化学寄りのバックグラウンドの人
- まずは分子構造データや物性データを表形式で整理し、簡単な統計・可視化から始めるとよいです。
- 記述子や分子フィンガープリントの概念を押さえると、「構造を数字で見る」感覚がつかめます。
- 機械学習を学ぶ際も、「どの構造がどう誤分類されるか」を化学的に眺めてみると理解が深まります。
データサイエンス寄りのバックグラウンドの人
- 原子・結合・官能基といった基本的な化学の用語と、SMILES や構造式の読み方を押さえるところから始めると、問題設定が見えやすくなります。
- 「分子はグラフ」「反応はグラフ変換」という視点で捉えると、アルゴリズム的な直感が活かせます。
- 一般の表形式データと違い、「同じ特徴量でも意味が違う」「対称性や同型性を意識する」といったポイントがあることに注意すると良いです。
どちらにも共通すること
- どの問題でも、「どの程度の精度なら実務的に意味があるか」「どの領域で外しても許されるか」を意識することが大切です。
- モデルが出した答えをそのまま信じるのではなく、化学・安全性・コストの観点から人間がチェックする前提で設計すると、現実的なシステムになります。
まとめ
- ケモインフォマティクスは、化学構造や反応データを情報として扱い、物性予測や分子設計を支援する分野。
- 創薬・材料設計などで、実験前の候補絞り込みや設計空間の可視化に広く使われている。
- データの質・バイアス・化学空間の広大さ・合成可能性など、向き合うべき課題も多いが、深層学習や生成モデル、実験自動化との連携によって可能性が広がりつつある。
- 「化学の知識 × データサイエンス」の掛け合わせが力を発揮する分野であり、どちら側のバックグラウンドから入る人にも学び甲斐のある領域と言える。
本記事が、「ケモインフォマティクスって何をしているのか?」をざっくり掴むための入口になれば幸いです。
ご自身の興味や専門と重ね合わせて、「どこから関わると面白そうか」を考えるきっかけになればうれしいです。