Seurat version5 (v5) のアップデートに伴い、以下の 3 つの関数が一つにまとめられた。
・SelectIntegrationFeatures
・FindIntegrationAnchors
・IntegrateData
・(PrepSCTIntegration)
→ IntegrateLayers
更に、主成分分析(PCA) を統合前に行うようになり、"integrated" アッセイも消失した。
とても軽量で単純になり、ユーザーの使用感が上がった一方、オブジェクトの構造が見えにくくなってしまったように感じて、少しだけ悲しい。オブジェクト理解は、解析を行う上で重要だと思う。
また、これは以前からだが、各関数がオブジェクト内のどこを参照しているのかについては、ほとんど言及されていない。これも少し悲しい。
そこで今回は、 v4 integration と v5 integration の良し悪し、および IntegrateLayers 関数導入に伴うオブジェクト構造変化を考えていく。注意点として、v4 integration は Seurat v5 でも機能しており、今回の記事は Seurat v5 における比較検証である。
先に v4 integration と v5 integration の良し悪しについて、結論だけ。
・コードの記載やパラメータ吟味を考えると、v5 の方が少し楽。
・v5 の方がバッチを強力に修正するという結果が観察されたが、サンプル間の生物学的多様性も失っている可能性がある (v4 と両方試したほうが良いのではないか)
・容量圧迫は v5 の方が少なくて良い。
・オブジェクト構造は v4 の方が把握しやすい。
まずはダウンロードしたファイルの整列から。.tar ファイルの解凍は割愛。
# GSE174481_RAW.tar を解凍し、解凍後の大量のファイルを下記のディレクトリに格納し、ディレクトリ移動
dir <- "/path/to/my_dir"
setwd(dir)
# サンプル名の識別
samples <- unique(substr(list.files(), 1, 10))
samples <- samples[grep("^GSM", samples)]
# Read10X 用にフォルダ作成、データ成形(この操作は1回だけでOK)
for(sample in samples){
# file 名を取得
files <- list.files(pattern = sample)
# folder 作成
dir.create(sample)
# file copy のためのパス定義
files_from <- paste(dir, files, sep = "/")
files_to <- paste(dir, sample, c("barcodes.tsv.gz", "features.tsv.gz", "matrix.mtx.gz"), sep = "/")
# file copy
file.copy(from = files_from, to = files_to)
# file remove
file.remove(files)
}
下記のようなフォルダが並んでいれば OK。
barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz がそれぞれ格納されている。
では v4 の integration を考える。v4 では、リスト型のオブジェクトが integration で使用されていたため、lapply 関数が結構役に立っていた。lapply 関数は、リスト内のオブジェクトにそれぞれ同じ操作ができ、結果を同様にリストで返す。dplyr のパイプ演算子を利用すると、更に楽になる。
解析を段階に分けるなら、以下のようなコードだ。なお、コードは v5 でも使用できる。
# R のバージョン: 4.4.2
library(Seurat) # 5.3.0
library(sctransform) # 0.4.2
library(glmGamPoi) # 1.16.0
library(dplyr) # 1.1.4
# Read10X 関数による読み込み(リストにすることで、後で楽になる)
object.list <- list()
use_samples <- samples[1:7] # 培養細胞サンプルの除外
for(sample in use_samples){
object.list[[sample]] <- Read10X(sample)
object.list[[sample]] <- CreateSeuratObject(object.list[[sample]], project = sample)
object.list[[sample]][["percent.mt"]] <- PercentageFeatureSet(object.list[[sample]], pattern = "^MT-")
}
# QC (あらかじめ閾値は決めてあるものとし、今回は一律で。)
object.list <- lapply(object.list, FUN = function(x){
x <- subset(x, subset = nFeature_RNA > 2500 & percent.mt < 10)
})
# SCTransform 正規化
object.list <- lapply(object.list, FUN = function(x){
x <- SCTransform(x, method = "glmGamPoi", vars.to.regress = "percent.mt", verbose = TRUE)
})
# Integration(正準相関分析以降に使用する次元は、適当に 15 とした。)
features <- SelectIntegrationFeatures(object.list = object.list, nfeatures = 3000)
combined_v4 <- object.list %>%
PrepSCTIntegration(anchor.features = features) %>%
FindIntegrationAnchors(anchor.features = features, dims = 1:15, normalization.method = "SCT", reduction = "cca") %>%
IntegrateData(normalization.method = "SCT",dims = 1:15)
# PCA → Clustering → UMAP(上記同様パラメータは適当。)
combined_v4 <- combined_v4 %>%
RunPCA(verbose = FALSE, npcs = 50) %>%
FindNeighbors(reduction = "pca", dims = 1:30) %>%
FindClusters(algorithm = 3, n.iter = 100, resolution = 0.5) %>%
RunUMAP(reduction = "pca", dims = 1:30)
パラメータ吟味を行うならば、QC 以降は以下のようなまとめコードも良いかもしれない。今回は記載しないが、for ループを加えれば、パラメータを変えながら一度で吟味を行うことが出来る。
# QC, SCT
object.list <- lapply(object.list, FUN = function(x){
x <- subset(x, subset = nFeature_RNA > 2500 & percent.mt < 10)
x <- SCTransform(x, method = "glmGamPoi", vars.to.regress = "percent.mt", verbose = TRUE)
})
# Integration
features <- SelectIntegrationFeatures(object.list = object.list, nfeatures = 3000)
combined <- object.list %>%
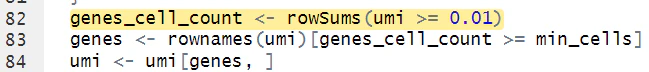
PrepSCTIntegration(anchor.features = features) %>%
FindIntegrationAnchors(anchor.features = features, dims = 1:15, normalization.method = "SCT", reduction = "cca") %>%
IntegrateData(normalization.method = "SCT",dims = 1:15) %>%
RunPCA(verbose = FALSE, npcs = 50) %>%
FindNeighbors(reduction = "pca", dims = 1:30) %>%
FindClusters(algorithm = 3, n.iter = 100, resolution = 0.5) %>%
RunUMAP(reduction = "pca", dims = 1:30)
続いて v5。こちらは QC 以降のまとめコードだけ記載するが、特にSelectIntegrationFeatures 関数が無くなったことで、 1 コードで済むようになり、とても分かりやすくなった。PCA を Integration 前に行っている点だけ注意。
combined_v5 <- merge(object.list[[1]], object.list[-1]) %>%
subset(subset = nFeature_RNA > 2500 & percent.mt < 10) %>%
SCTransform(method = "glmGamPoi", vars.to.regress = "percent.mt", verbose = TRUE) %>%
RunPCA(npcs = 50) %>%
IntegrateLayers(method = CCAIntegration, orig.reduction = "pca", new.reduction = "integrated.cca", normalization.method = "SCT", verbose = FALSE) %>%
FindNeighbors(reduction = "integrated.cca", dims = 1:30) %>%
FindClusters(algorithm = 3, n.iter = 100, resolution = 0.5) %>%
RunUMAP(reduction = "integrated.cca", dims = 1:30)

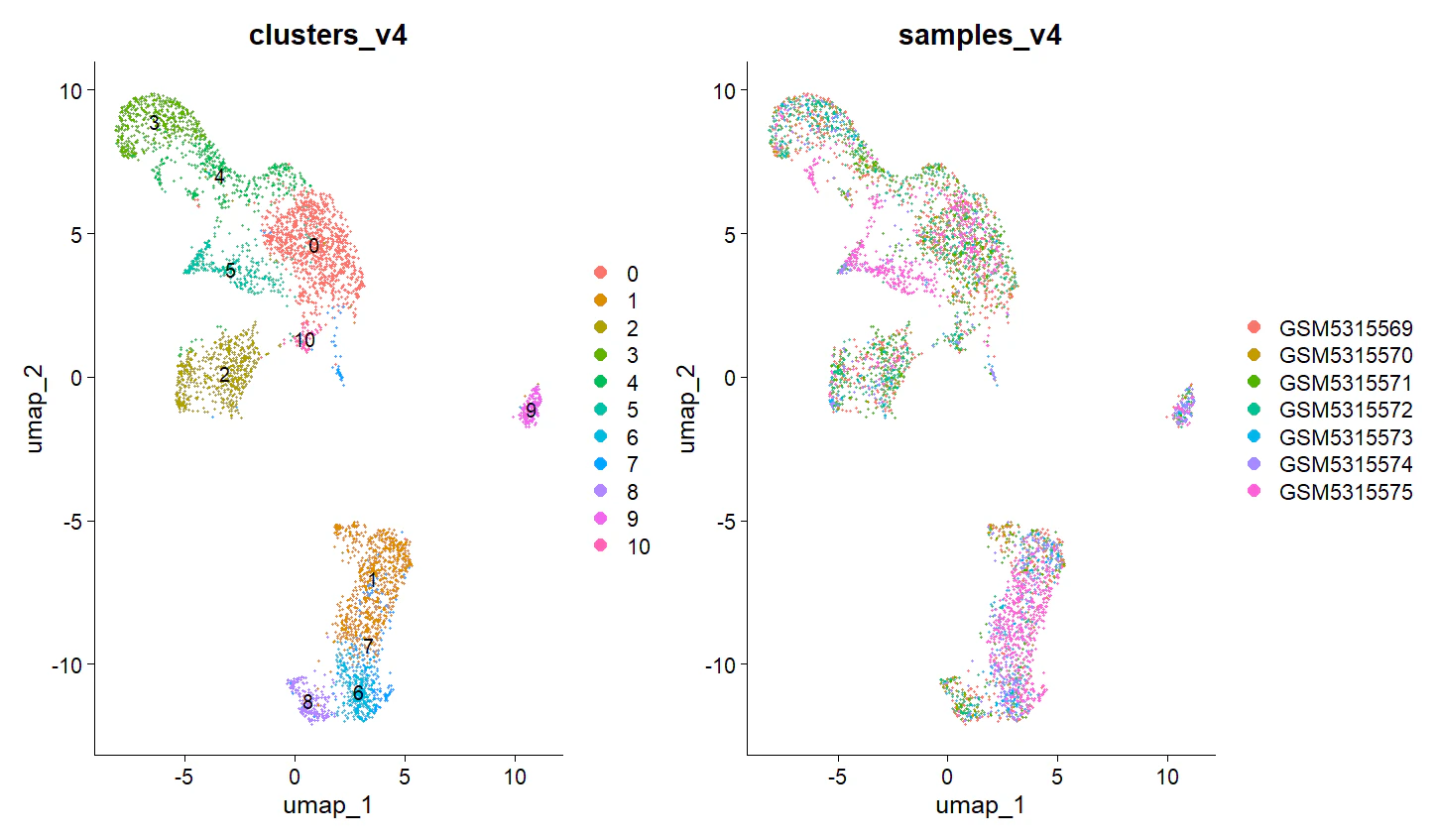
ちなみに、v5 UMAP 上で v4 クラスタリング結果を反映したものが以下だ。

上図の下部は線維芽細胞なのだが、v4 では分かれていたものがまとまっている。この点に関しては以下に議論があるが、明確な答えは得られていない。
https://github.com/satijalab/seurat/issues/9494
可能性として、この現象は PCA のタイミングが関係するのではないかと考えている。v5 では PCA が統合前に行われることにより、サンプル間の大局的な構造の違い (つまりバッチ) が大きく修正されているのではないだろうか。逆に言えば、v4 ではバッチがノイズとして最後まで残っている可能性がある。実際、記事の上の方で示した v4 の図では、クラスター 1,6,7,8 の別れ方とサンプルの別れ方に類似性があるように見える。一方、v5 では特に、サンプル間の生物学的多様性をバッチとしている可能性もあるため、どちらが良いと判断することは難しい。どっちも試すのが一番だろう。
また、容量的な意味では、v5 の方が少ない。これは恐らく、integrated assay の消失が大きく関係している。詳しい話は、以下のオブジェクト構造の比較で説明する。

ここまでで、大まかな比較はできた。次は、内部構造の話に入っていく。必要ならば、以下も参照して欲しい。
https://qiita.com/Refine/items/e92c4a62fb23e9ed87f0
また、前提として、SCTransform が NormalizeData, FindVariableFeatures, ScaleData の役割を果たす関数であることは抑えておいて欲しい。SCT の詳しい理論考察は以下。
https://qiita.com/Refine/items/c58d9cb9814e33a1092f
では、v4, v5 integration における構造の違いをみていく。方法としては、identical 関数を用い、TRUE にならない部分を全て見ていった。まずは SCTransform 前後の違いについて記載しておく。
RNA assay に変化は無いようだ。

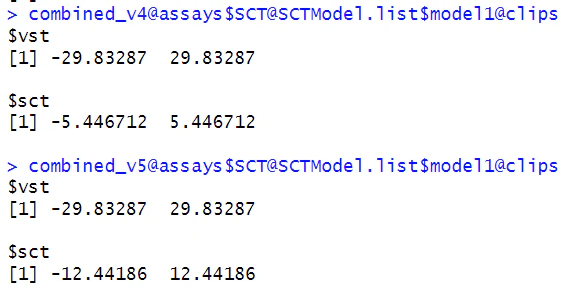
SCT におけるクリッピング √(N/30) の値が変わっている。v5 では、全細胞数が参照されているようだ。恐らく PCA への入力時にクリッピング値の差が出ることを防ぐための変更だと思われる。ちなみに、GSM5315569 は 890 細胞なので、sqrt(890) ≒ 29.83, sqrt(890/30) ≒ 5.44 である。また、全細胞数は 4644 なので、sqrt(4644/30) ≒ 12.44 である。
SCT 後に得られるピアソン残差 (scale.data) の行列数が変わっている。ちなみに、v4 の 3000 に関しては、SelectIntegrationFeatures 関数で選択された 3000 遺伝子と一致する(順番は異なる)。

この違いについてだが、v5 側の SCTransform 関数を debug している途中に、vst 関数内に以下の 2 コードを見つけた。これは、各 SCT model の作成時に保持されている VariableFeatures の和集合 (6408遺伝子) と、画像に示すような、発現が 0 でない細胞が min_cells(=5) 個以上である遺伝子の積集合 (12489遺伝子) の、積集合(5357)遺伝子として表される。和集合に関しては恐らく、PCA で構造を失わないように各サンプルの変動遺伝子をなるべく保持したいという狙いがあるのだろうが、最初の積集合に関してはよく分からない。全く異なるサンプルや細胞型が含まれる場合に、影響が出そうな気がするが...(v4, v5 の違いに影響?)
if (return.only.var.genes) {
var_features_union <- Reduce(union, lapply(output_list,
function(output) {
return(VariableFeatures(output))
}))
all_features_intersect <- Reduce(intersect, lapply(output_list,
function(output) {
return(rownames(output))
}))
scale_data_features <- intersect(all_features_intersect,
var_features_union)
}
ちなみに、counts, data に関しては sct.clip が行われるよりも前の段階で計算されるため、v4, v5 で全く同じ結果を返す。
また、v5 だけ SCT assay の var.features に遺伝子名が格納されている。
次に、integration について。v4 では統合時に SCT assay 内の scale.data を参照していたが、これは v5 でも同様のようだ。加えて、v5 では PCA のcell.embeddings (主成分得点) がどうやら奥深くの PairwiseIntegrateReference 関数内で使用されているようである。残念ながら、詳細な理論までは把握出来ていない。

最終的に、v5 では根本的に "integrated assay" が消え、reductions の部分に追加されている。
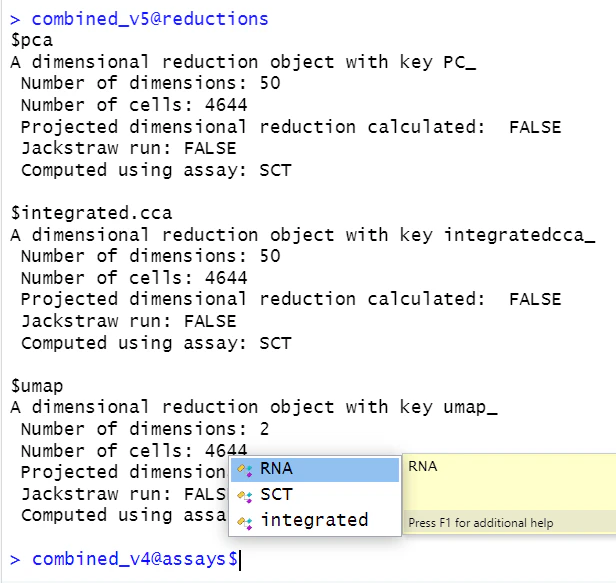
それに伴い、各関数が参照する場所も変化している。例えば、v4 では、FindNeighbors, RunUMAP はともに PCA の cell.embeddings を参照していたが、v5 では、integrated.cca の cell.embeddings を参照している。

v5 の少し難しいと感じる点は、PCA が統合前に行われている点と、integrated assay が消えてしまった点にある。v4 では正規化 → 統合 → PCA → UMAP and cluster 解析という流れで、正規化と統合はそれぞれ assay としてまとめられていた。そのため、統合の部分で assay としての区切りを意識づけしやすかった。一方 v5 では、正規化 → PCA → 統合 → UMAP and cluster 解析 という流れになったことで、統合の意味を次元削減の一種として捉えることとなり、流れの切れ目が見えにくくなった。これにより、少し全体像が見づらくなったように感じる。
個人的にはどちらも使用するつもりだが、慣れないうちは v4 を使う方が分かりやすいかもしれない。
次回は、integration の理論を見ていこうかな...