こんにちは。Seurat、楽しんでますか?
今回は、Seurat を始めたての人に向けて、最新: version 5.3.0 時点における Seurat Object の構造と中身の参照方法を、基本的な流れを解説しつつ紹介します。
※ この記事は、Seurat のチュートリアルを一通り読んでいることを想定しています。基本的な R に関する用語は解説しませんので、適宜必要に応じて調べてください。また、以下では "@", "$" のような参照方法が頻発しますが、それぞれの意味はここでは割愛します。どっちも良く使うので、今は二つあることだけ常に頭に入れておけば大丈夫です。
使用パッケージ
Seurat: 5.3.0
SeuratObject: 5.1.0
まず、データを RStudio に読み込みます。
データは以下より入手し、windows の 7-zip で解凍しました。mac terminal でも、gzip, tar コマンド辺りで解凍できると思います。("here" よりダウンロード)
https://satijalab.org/seurat/articles/pbmc3k_tutorial.html
「filtered_gene_bc_matrices」というフォルダがあると思いますが、大事なのは中にある .tsv, .mtx もしくは .tsv.gz, .mtx.gz です。どのフォルダに保存するかは自由ですが、必ず名前を変えず、セットでおいてください。

データ読み込みのコードは以下のようになります。data.dir は、現在のディレクトリが一致していれば相対パスでも大丈夫です。今いるディレクトリが分からない場合は、getwd() で確認してください。なお、下記の assay = "RNA" はデフォルトですが、後ほど重要な意味を持ちます。project = "sample" については、サンプル識別のようなものと考えて大丈夫です。
library(Seurat) # version 5.3.0
object.data <- Read10X(data.dir = "~/filtered_gene_bc_matrices/hg19")
object <- CreateSeuratObject(counts = object.data, assay = "RNA", project = "example")
この他にもファイルの形式によって、様々な読み込み方法があります(.h5, .h5ad, .h5Seurat, .rds, ect...)。ここでは解説しませんが、必要になったときに学ぶ程度で良いと思います。
では、object の中身を見ていきます。
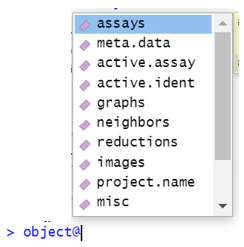
① assays: 解析の根幹データである、遺伝子数(行)×細胞数(列)の行列が格納されています。assays の中には現状 "RNA" のみが表示されています。
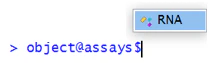
以下は "RNA" の中身です。Seurat version 5.0 から、カウント行列(layer)、細胞名(cells)、遺伝子名(features) が別々に格納されるようになりました。それ以外の項目は特に気にしなくていいと思います。

エクセルのような視覚的にわかりやすい情報として行列を取得したい場合には、以下のコードを活用してください。なお、"counts" は layers 内にあり、生のカウントを指します。

matrix <- GetAssayData(object, assay = "RNA", slot = "counts")
先ほど、assay = "RNA" が重要だと述べましたが、主な理由は解析手法により、同一名の行列データが複数種類存在することがあるからです。例えば "counts" は、正規化前か、正規化後 (SCTransform) か、統合後 (integration) かという違いがあるため、重複しないよう assay を分ける必要があります。assay 名はそれぞれ "SCT", "integrated" とされます。ちなみに、Visium 空間トランスクリプトームでは、デフォルトの assay 名は、"RNA" ではなく "Spatial" です。
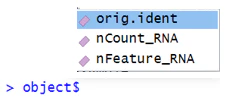
② meta.data: 最初に "example" として指定したサンプル名(orig.ident), 細胞ごとのシーケンス深度 (nCount_RNA), 細胞ごとの検出遺伝子種類数 (nFeature_RNA) がデフォルトで記載されています。

また、meta.data に関しては、以下のようにショートカットして中身を見ることもできます。
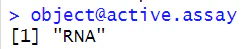
③ active.assay: そのままの意味ですが、現在どの assay を参照しているかを指します。SCTransform, integration を行った際は、注意すべき点だと思います。同じ "counts" でも、違う assay が参照されていたなんてことは、よくあります。
また、active.assay を変更することもできます。ここでは RNA assay しかありませんが、一応。
DefaultAssay(object) <- "RNA"
④ active.ident: 細胞ごとの ident に何が使用されているかが記載されています。例えば、クラスター解析の後だと、クラスター番号のラベルが付けられます。細胞ごとのラベルを色々変えたい人にとっては、あまり必要のない項目だと思います(メタデータに記載しておけば毎回参照できるため)。
では、一旦ここまで見たところで、1 サンプル解析パイプラインを流してみます。ただし、正規化には SCTransform を使用します。SCTransform の解析理論は以前の記事を参照してください。https://qiita.com/Refine/items/c58d9cb9814e33a1092f
また、QC の閾値や次元の決め方については、tutorial を参照してください。
# meta.data に、死細胞の指標であるミトコンドリア由来遺伝子のカウント割合を付加します。解説はコメントアウトとして記載します。
# pattern = "^MT-" は正規表現で、MT- から始まる遺伝子を指します。
object[["percent.mt"]] <- PercentageFeatureSet(object, pattern = "^MT-")
# QC を行います。
object <- subset(object, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
# SCTransform 正規化
object <- SCTransform(object, vars.to.regress = "percent.mt", verbose = FALSE)
# 線形次元削減(Principal Component Analysis: PCA)
# デフォルトで 50 次元を出力します。
object <- RunPCA(object, verbose = FALSE)
# 非線形次元削減(Uniform Manifold Approximation and Projection: UMAP)
# 今回は PCA で得られた次元のうち、30 次元を使用します。
object <- RunUMAP(object, dims = 1:30, verbose = FALSE)
# Annoy による K-Nearest Neighbors(KNN) の計算と、Jaccard Index (Shared Nearest Neighbors: SNN) の計算
# 使用する次元数は、UMAP と統一します。
object <- FindNeighbors(object, dims = 1:30, verbose = FALSE)
# Louvain Algorithm によるクラスター解析
object <- FindClusters(object, verbose = FALSE, resolution = 0.8)
# 一応可視化
DimPlot(object, label = T) + NoLegend()
UMAP 上の向きの違いはあれど、tutorial と概ね一致する結果が得られました。

ここから先ほどの続きを見ていきますが、その前に、SCTransform やクラスター解析によっていくつか追加されている内容があります。確認してみてください。
※ assay に SCT が追加

※ meta.data に以下が追加。
nCount_SCT, nFeature_SCT は、 RNA assay と計算方法は同じです。
SCT_snn_ews.0.8 は、FindClusters 関数の resolution に対応するクラスター番号です。
seurat_clusters は、現在の resolution に対応するクラスター番号です。(resolution を変えて再解析していないので、SCT_snn_ews.0.8 と一致します)。

※ active.assay が SCT に変更。

※ その他、active.ident が、クラスター番号に変更。
ただし、RenameIdents 関数を使えば、active.ident は変更可能です。
では、続きを見ていきます。
⑤ graphs: KNN, SNN の結果が記載されています。中身を見ればわかりますが、細胞数×細胞数の行列になっています。SCT_nn は、近傍なら 1, 近傍でないなら 0 を示す行列で、自身は 1 です。SCT_snn は、KNN より計算された Jaccard Index です。

⑥ neighbors: 以前はここに KNN の結果が格納されていた気がするのですが、もしかすると廃止されたのかも?
⑦ reductions: 次元削減の結果が格納されています。重要なものだけピックアウトします。※ 主成分得点の計算は飛ばしても大丈夫です。

cell.embeddings はいわゆる座標を表します。PCA では、軸ごとの主成分得点にあたります。feature.loadings は、PCA における固有ベクトルで、UMAP では空です。sdev は、PCA における固有値の平方根で、UMAP では空です。
つまり、主成分得点は以下のように計算が出来ます。
# 主成分得点
point <- object@reductions$pca@cell.embeddings
# 固有ベクトル
load <- object@reductions$pca@feature.loadings
# SCTransform により得られたピアソン残差の転置
mat <- t(object@assays$SCT@scale.data)
# 遺伝子名の整列
mat <- mat[,rownames(load)]
# 行列積
point2 <- mat%*%load
# 確かめ(小数第8位までは一致を確認)
identical(round(point, digit = 3), round(point2, digit = 3))
⑧ images: Visium では画像情報が入っていますが、scRNA-seq では空です。
⑨ project.name: CreateSeuratObject 関数の "example" です。
⑩ その他、version は恐らく SeuratObject パッケージの versionを示します。 misc, tools は不明ですが特に使用したことは無いです。commands については、コマンドの履歴を見ることが出来ます。しかし、僕はこれをみてデータを再現できたことは無いです。
以上となります。いかがだったでしょうか。オブジェクトの構造把握は大きな障壁になりやすく、生成 AI に頼っていっても中々身に付かない情報です。慣れていくことで、Seurat をよりエンジョイして貰えたらなと思います。
次回は、KNN, SNN の理論を書いていくか、integration で今回のように細かい部分を書いていくか、悩みどころです。