この記事はバイオインフォマティクス Advent Calendar 2019の21日目の記事です。
なぜワークフローエンジン?
- あれこれのツールや、自作のスクリプトがたくさんあって、それぞれてんでばらばらな入出力のとりかたをする
- シェルスクリプトで全体をつないでやることにする
- あれこれデータを置くディレクトリ構造やファイル名を管理して上書きしたりしないようにする
- ファイルのキャッシュ機能とかも自分で実装する
- だんだんややこしくなってくる
- 一から作り直したくなる
こんなしんどさを経験される方は多いのではないでしょうか。そういうつらさをすっきり解消してくれる(希望的観測)のがワークフローエンジンです。今回はその一つであるNextflowを初歩の初歩からちょっとだけご紹介します。レベルの高い方々には物足りない内容となりますがご勘弁ください。
Nextflowとは
NextflowはバルセロナのCentre for Genomic Regulationによってオープンソースで開発されている科学計算用ワークフローエンジンです。GitHubのスター数はおよそ1000、現在かなり活発に開発が進められています。Java上で走るGroovyで書かれていて、可搬性、再現性、スケーラビリティなどを売りにしています。Nextflowについてはすでに NextflowからCWLで書かれたワークフローを呼び出す というやや応用的な記事が@yuifuさんによって書かれていますが、本記事はより初歩的な内容をカバーしています。ただ、そもそもどのような種類のワークフローエンジンがあるかについてもまとめてくださっているので、本記事を読まれる前に前半だけでも読まれることをおすすめします。また@manabuishiirbさんによる Nextflow を使ってみる と@shiromadaraさんによる コアファシリティのゲノムインフォ技術員の仕事を楽にしてくれるツール等の紹介 もあわせてご覧ください。
インストールと既存パイプラインの実行
超簡単! インストールしたい先で curl -s https://get.nextflow.io | bash するだけです。単一バイナリの nextflow が現れます。それ以外に %HOME/.nextflow が生成されてあれこれのファイルが置かれますが、触る必要はありません。モダンな設計で素敵ですね。なおJava8以降が必要です。
どこかパスが通っているディレクトリにインストールできたら nextflow run hello でHello Worldしてみましょう。
> nextflow run hello
N E X T F L O W ~ version 19.10.0
Pulling nextflow-io/hello ...
downloaded from https://github.com/nextflow-io/hello.git
Launching `nextflow-io/hello` [maniac_brattain] - revision: a9012339ce [master]
WARN: The use of `echo` method is deprecated
executor > local (4)
[18/df5059] process > sayHello (4) [100%] 4 of 4 ✔
Bonjour world!
Ciao world!
Hello world!
Hola world!
実際にはこれは手元にあるパイプラインを実行しているわけではなく、 https://github.com/nextflow-io/hello を取得&実行しています。細かい設定は何もしなくても、 nextflow run foo/bar で http://github.com/foo/bar から取ってきてくれます。さらに -user オプションでprivate repositoryからの取得もできたり、 -r オプションでバージョン指定もできます。GitHub以外からの取得にも対応しています。詳細は Pipeline sharing 。
自分でパイプラインを作ってみる
まずは Get started の通りのものを走らせてみましょう。以下の内容を tutorial.nf として保存します。
# !/usr/bin/env nextflow
params.str = 'Hello world!'
process splitLetters {
output:
file 'chunk_*' into letters
"""
printf '${params.str}' | split -b 6 - chunk_
"""
}
process convertToUpper {
input:
file x from letters.flatten()
output:
stdout result
"""
cat $x | tr '[a-z]' '[A-Z]'
"""
}
result.view { it.trim() }
その上で nextflow run tutorial.nf すると次の出力が得られます。
N E X T F L O W ~ version 19.10.0
Launching `tutorial.nf` [cranky_nightingale] - revision: e3b475a61b
executor > local (3)
[e9/ffdfbd] process > splitLetters [100%] 1 of 1 ✔
[8f/8635ef] process > convertToUpper (2) [100%] 2 of 2 ✔
HELLO
WORLD!
実行した tutorial.nf の中身を見てみましょう。細かい文法は抜きにして、 Hello world! というもともとの文字列が process splitLetters によって単語に分割され、それが process convertToUpper で大文字に変換されて出力されていることがなんとなくわかります。また """ で囲まれている範囲はシェルスクリプトっぽいこともわかりますね。
さて、決め打ちされた入力を処理するだけではつまらないですから、引数を与えてその内容を処理してもらいましょう。実はこのパイプラインに何も変更を加えなくても実現可能です。 nextflow tutorial.nf --str "bioinformatics" を実行してみてください。
N E X T F L O W ~ version 19.10.0
Launching `tutorial.nf` [distracted_lichterman] - revision: e3b475a61b
executor > local (4)
[9d/571802] process > splitLetters [100%] 1 of 1 ✔
[fd/e18b6c] process > convertToUpper (2) [100%] 3 of 3 ✔
BIOINF
CS
ORMATI
このように、 params.str の内容は --str で指定、というふうに引数を与えて処理させることができます。
ここまでに気づいた方もいると思いますが、分割された文字列の出力順は一意に定まりません。これは convertToUpper が並列実行されるためです。
Processとchannelによるデータのハンドリング
Basic concepts や Processes あたりの内容。ここでprocessと言った場合には一般的な意味(スレッドとプロセスという意味でのプロセス)ではなく、Nextflow上での実行単位としての意味であることに留意してください。
Nextflowにおいては、processが独立した一つの処理単位となります。異なるprocess間では状態は共有されません。唯一process間がやりとりできるのは、channelを介してだけです。
こうすることによって、process間での依存関係が明確になり、何も指示しなくても適切な順番でprocessを実行してくれます。さらに、複数のprocessのうち一部が変更された場合に、変更箇所とその下流だけ再実行し、変更していない箇所は以前の結果のキャッシュを使いまわすことも自動的に行ってくれます。
それぞれのprocessが実際に何を実行するかは、上で見たようなBash scriptとして記述されます。システム上にあるものならどんなコマンドも使えますし、パイプしたり複数行にわたって書くこともできます。注意点はシェル内での変数とNextflowが持っている変数がそれぞれあることです。そのあたりのハンドリングについては Processes を見てください。
そしてprocess間をつなぐ channel には二つの種類があります。
- Queue channel
- Value channel
Queue channelは一般的なqueueとして先入先出で要素を管理します。言い換えれば、要素を取り出せばなくなってしまうということです。対してvalue channelは単一の値を持ち、何回でも参照することができます。
そろそろ具体的に入出力があるケースを見てみましょう。以下はみんな大好きBLASTを実行する例です。
# !/usr/bin/env nextflow
/*
* Defines the pipeline inputs parameters (giving a default value for each for them)
* Each of the following parameters can be specified as command line options
*/
params.query = "$HOME/sample.fa"
params.db = "$HOME/tools/blast-db/pdb/pdb"
params.out = "./result.txt"
params.chunkSize = 100
db_name = file(params.db).name
db_path = file(params.db).parent
/*
* Given the query parameter creates a channel emitting the query fasta file(s),
* the file is split in chunks containing as many sequences as defined by the parameter 'chunkSize'.
* Finally assign the result channel to the variable 'fasta'
*/
Channel
.fromPath(params.query)
.splitFasta(by: params.chunkSize)
.set { fasta }
/*
* Executes a BLAST job for each chunk emitted by the 'fasta' channel
* and creates as output a channel named 'top_hits' emitting the resulting
* BLAST matches
*/
process blast {
input:
file 'query.fa' from fasta
file db_path
output:
file 'top_hits'
"""
blastp -db $db_path/$db_name -query query.fa -outfmt 6 > blast_result
cat blast_result | head -n 10 | cut -f 2 > top_hits
"""
}
/*
* Each time a file emitted by the 'top_hits' channel an extract job is executed
* producing a file containing the matching sequences
*/
process extract {
input:
file top_hits
file db_path
output:
file 'sequences'
"""
blastdbcmd -db $db_path/$db_name -entry_batch top_hits | head -n 10 > sequences
"""
}
/*
* Collects all the sequences files into a single file
* and prints the resulting file content when complete
*/
sequences
.collectFile(name: params.out)
.println { file -> "matching sequences:\n ${file.text}" }
ざっと流れを見ていきましょう。まず一通り変数を定義したあと、 fasta というchannelにsample.faを分割したものを流し込んでいることがわかると思います。
その上で、先に実行される process blast では fasta から一つ一つファイルを取り出して、 query.fa という名前で入力用に準備しています。ファイルの重複とか上書きとかが問題にならないように、一つ一つ分けた一時ディレクトリをNextflowが作って管理して、こうやって特定のファイル名の入力を用意するところまでやってくれるわけです。今回の blastp は入力ファイル名が可変なのでここまでの必要はないですが、特定のファイル名しか受け付けないツールを走らせるときに便利でしょう。もう一つの入力である db_path はこのprocess内ではいっさい実際のファイル名に関知しないままで与えることができています。出力ファイル名も同様に繰り返し実行されることを気にせずに与えています。
次に実行される process extract ではBLASTでヒットしたエントリをDBから取り出しています。ここでもファイル管理がまったくpainlessになっています。そして最後に結果をまとめて表示するところまでで完了です。
このように、たくさんのファイルがあって、あちこちからさまざまな入出力ファイルがあって、それが繰り返し実行される場合でも、あたかも一回だけ実行するかのように容易に扱えることがわかったと思います。なお、このときにメタデータをファイル名に残しておかないと何をどのように処理したものかがわからないという心配をされるかもしれません。Nextflowではそのような場合には tuple 型を用いてメタデータを別途持つことを推奨しています。
実践的に動かしてみる
上の例は動かしにくいので、もっと実践的にひとつやってみましょう。Awesome Nextflowにいろいろありますが、ここではkallistoをやってみることにします。kallistoとsleuth、それからRにbioconductorのbiomRartパッケージをインストールしておいた上で、 nextflow run cbcrg/kallisto-nf を実行してみてください。これだけでサンプルデータでの結果が /results 以下に出力されるはずです。
さらに、 nextflow run cbcrg/kallisto-nf -with-report とするとhtml形式でレポートが出力されます( Tracing & visualization )。
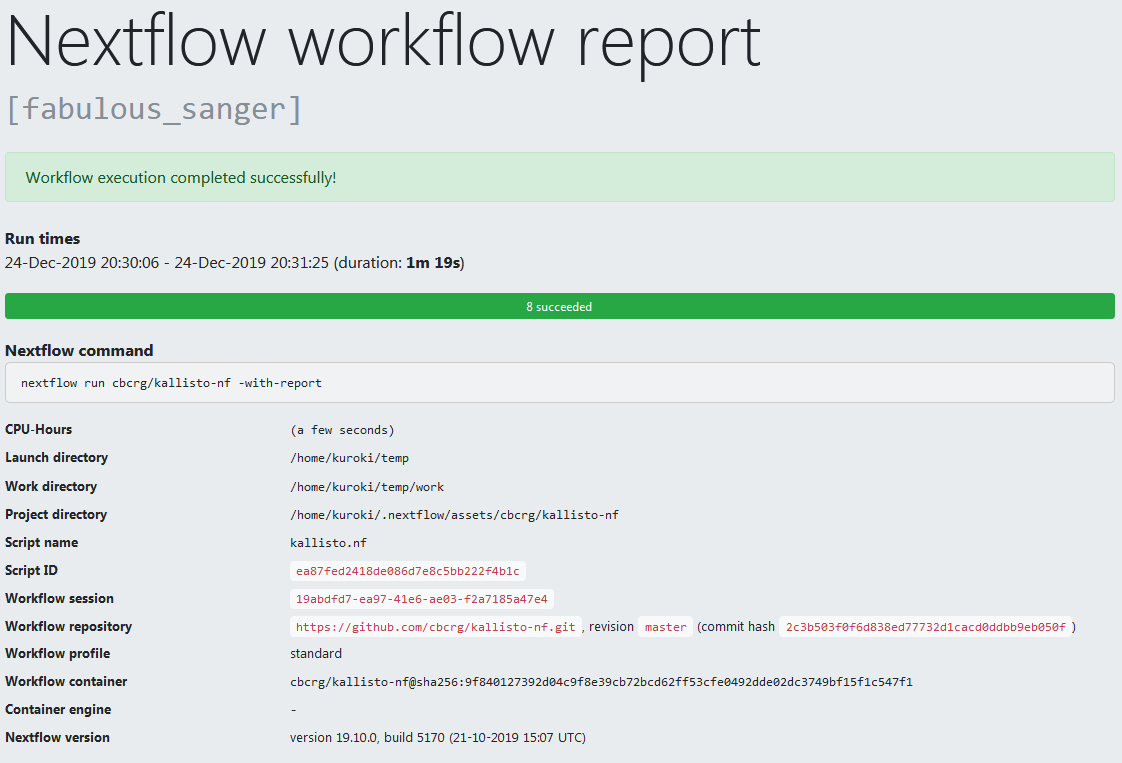
個別の実行結果もご覧のとおり。
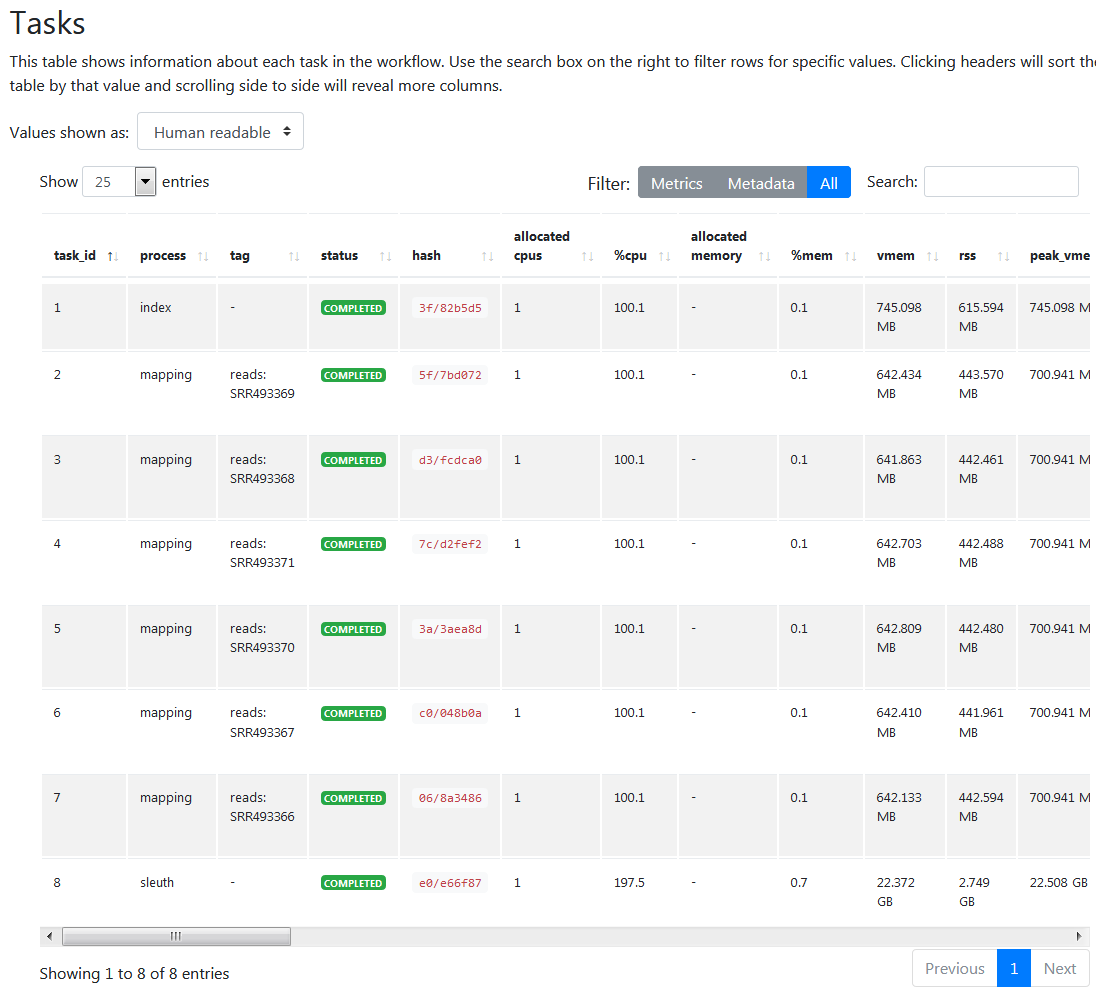
さらに、これをそのまま qsub で動かすことだってできます。github.com/cbcrg/kallisto-nfに説明があるとおり。あれこれやらなくてもたった数行の設定ファイルだけで簡単にできる……はずなのですが、qsubへの投入はされるものの、依存関係のインストールやら環境変数の設定やらで問題が出てきて正常に実行できていません。手間がかかりそうなのでこの点は後日に回すこととさせてください。cf. Executors
まとめ
いかがでしたでしょうか? ざっとこんなもので概要だけでもわかっていただけたら幸いです。おおむねチュートリアルをなぞっただけですが、少しだけ流れを加えた記事にしました。ここでは触れませんでしたが、NextflowはDocker/Singularityコンテナの実行やAWS、GCP上での実行などもサポートしています。ドキュメントにも記述があるとおり、NextflowはLinux環境がlinuga francaであるという前提のもと、その上でportabilityを高くする設計思想を持っています。POSIX準拠でシェルスクリプトを書くことに魅力を覚える方は、Nextflowの抽象性指向には似た雰囲気を感じるのではないでしょうか。その観点からすれば、Java上で動くGroovyを使ってprotabilityと表現力を両立しているのも納得がいくところです。このあたりはたとえばPythonエコシステム内で完結することを前提とするSnakemakeとの違いかと思います。いっぽう、CWLとの関係では、だいぶ記述量が少なくてすむ、プログラミング言語の機能をまるごと持っているのでなんでもできる、という点が違います。はじめは少しとっつきにくさもありますが、機能が多いながらもモダンかつシンプルな設計でかなり気に入りました。