AlphaFold3が化合物を取り扱えるようになりましたが、化合物の入力方法はSMILES記法またはUser-provided CCDのみとなっております。
基本的にはUser-provided CCDのほうが拡張性は高く、共有結合の設定なども可能ということで上位互換性があるのですが、もし予測したい化合物が他の分子と共有結合を形成しない場合はSMILES記法でもOKです。
多くの生化学者にとってはSMILES記法は簡単じゃありません。しかし大学であればChemDrawが用意されているなど、構造式を自前で書ける環境は多いので、そこからSMILESに変換してほしいものです。そんなTIPSをいくつか紹介します。
PubChemからSMILESを取得する
目的の化合物がPubChemに登録されている場合、その情報をPubChemから取得することができます。
例として、Antibiotic LL-AB 664の場合、2.1.4 SMILESのところにページを移動させると、そこに記述されています。


この化合物のSMILES記法はCN1C[C@H]([C@@H]2[C@@H](C1=O)N=C(N2)N[C@H]3[C@@H]([C@@H]([C@H]([C@H](O3)CO)OC(=O)N)O)N(C)C(=O)CN=CN)Oでした。
SMILESが正しいかを確認したい
HPCシステムズ株式会社のページにSMILESを2次元分子構造に変換するツールがあります。これを使うとその場でSMILESを化学構造式の図に変換してくれますので、これで確認しましょう。

ChemDrawからSMILESに変換する
ChemDrawがあれば、化合物を描いておいて、そこからEdit -> Copy As -> SMILESを選択すれば、それだけでSMILESに変換された化合物構造式を得ることができます。


これで構造式のSMILES表記がコピーされた状態になるので、適当なエディタの上で「貼り付け」をしてみましょう。

慣れてきたら、ショートカットキーを使うと良いでしょう。Macの場合はOption(⌥)+Command(⌘)+Cボタンです。
GaussViewからSMILESに変換する
量子化学計算ソフトウェアのGaussian 16の公式ViewerであるGaussViewからも、sdf形式のファイルを経由してSMILESに変換できます。
Gaussviewで構造を作成します。このとき、立体配置がわかるように作られていればOKで、構造最適化をする必要はないと思います。
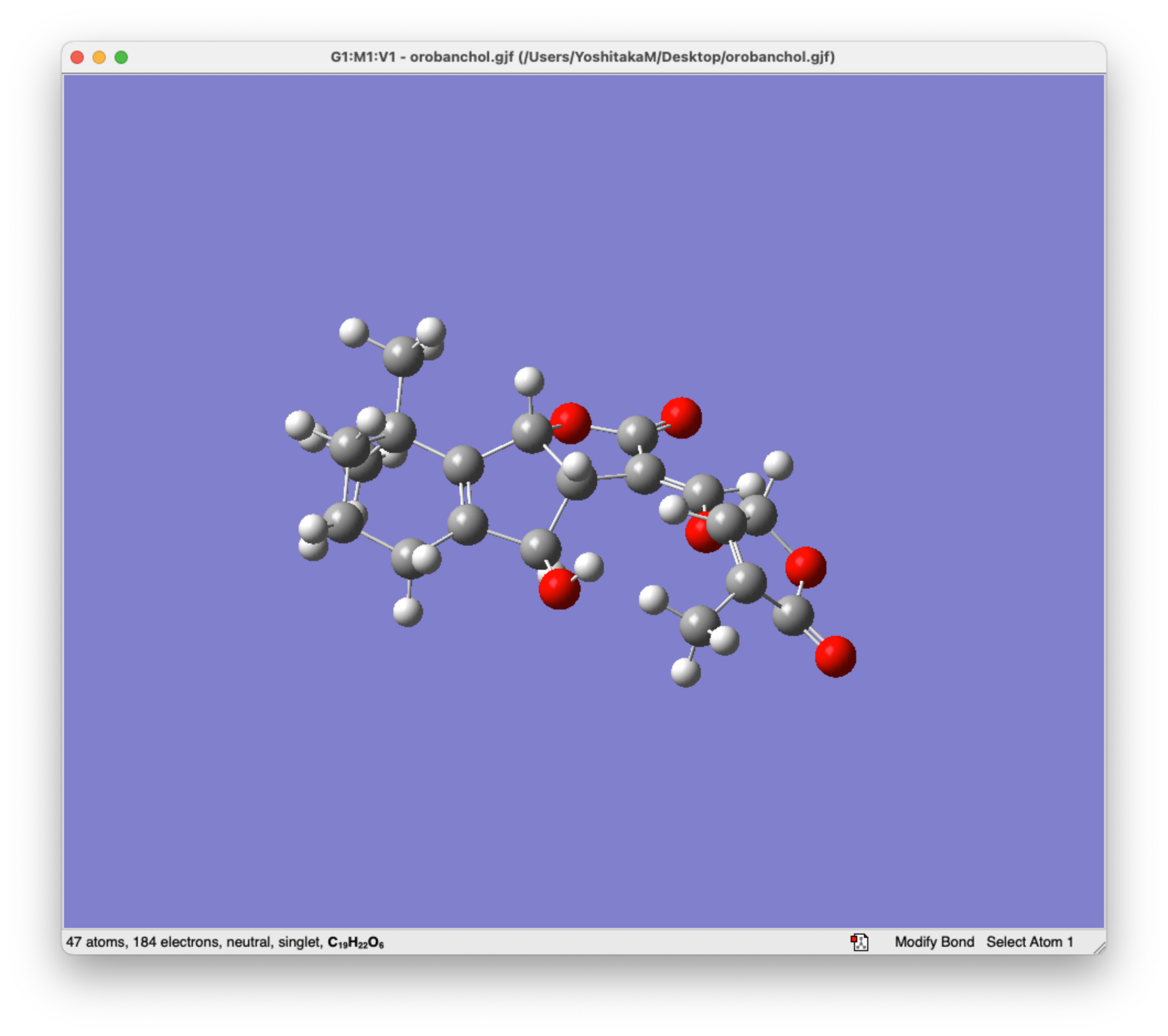
これを"File"->"Save"から"MDL SDF Files(.sdf)"で保存します。orobanchol.sdfというファイル名で保存されたとします。

このsdfファイルをRDkitを使ってSMILESに変換するのですが、このままrdkitで変換しようとすると失敗します。この時点のsdfファイルをテキストエディタで開くと
Created by GaussView 6.1.1
54 55 0 0 0 0 0 0 0 0 0 0
となっていますが、これを
Created by GaussView 6.1.1
54 55 0 0 0 0 0 0 0 0 0
となるように、最後の0を消してあげる必要があります。
続いて、RDKitでこれをSDFファイルからSMILES記法に変換します。
適当なディレクトリを作成し、その中で仮想環境を作成してrdkitをインストールして実行するのが適切でしょう。
# Homebrewでpython3.14をインストールしておく
brew install python@3.14
# 作業ディレクトリtest1を作成
mkdir -p ~/Desktop/test1
cd ~/Desktop/test1
# venvで仮想環境の作成
python3.14 -m venv .venv
# 仮想環境にrdkitをインストール
.venv/bin/python3.14 -m pip install rdkit
終わったら、作成した~/Desktop/test1ディレクトリにorobanchol.sdfファイルを入れて、以下のコンバータスクリプトsdf_to_smiles.pyを作成します。
from rdkit import Chem
# 化合物名を入力
sdffile="orobanchol.sdf"
supplier = Chem.SDMolSupplier(sdffile)
for mol in supplier:
if mol is not None:
smiles = Chem.MolToSmiles(mol, allHsExplicit=False)
print(smiles)
これでスクリプトを実行すると、SMILES表記が出力されます。
$ .venv/bin/python3.14 sdf_to_smiles.py
[12:16:06] Warning: molecule is tagged as 2D, but at least one Z coordinate is not zero. Marking the mol as 3D.
CC1=C[C@H](O/C=C2/C(=O)O[C@H]3C4=C(CCCC4(C)C)[C@H](O)[C@@H]23)OC1=O
というわけでCC1=C[C@H](O/C=C2/C(=O)O[C@H]3C4=C(CCCC4(C)C)[C@H](O)[C@@H]23)OC1=Oが得られました。
AlphaFold3ではアミノ酸・核酸も含めて、化合物について水素原子の情報を無視します。上のスクリプトでもallHsExplicit=Falseとしているのはこのためです。これゆえに、SMILES作成時には化合物を構成する原子の間に存在する結合価数が重要となります。一部の化合物については水素原子の有無が重要となるかもしれませんが、これを取り扱うことはできません。
Gaussian 16で化合物の構造を作成しているとき、化合物を構成する原子の間の結合価数は正しく設定する必要があります。また、共鳴構造を持つような化合物の結合が存在する場合(例えばCOO-など)、点線結合は使うことができません。例えば酢酸CH3COO-の場合

これを変換すると、SMILESはCC(:O):Oとなりますが(:が共鳴を表す)、これをAlphaFold3のインプットに加えた場合エラーが発生します。
この場合、Gaussian 16の中でC=O二重結合とC-OH単結合を設定してください。