この記事は創薬Advent Calendar 2018の4日目の記事です。
今回は、2017年のScience 355 (6322), pp. 294–298 に掲載された、David Baker研究室からの論文**Protein structure determination using metagenome sequence data**に使われていた手法のまとめとともに、実際にその効力を試してみるところまで紹介します。
創薬Advent Calendar 2018に書いておきながら、これはStructure-based drug designに役立てたらいいな〜くらいのノリで、直接創薬につながるというわけではありませんが、まあタンパク質構造の予測に興味のある方は読んでいってください。
Introduction
タンパク質の配列からの構造予測
タンパク質は20種類のアミノ酸でできています。これらがペプチド結合を介して数珠つなぎになったものはポリペプチドと呼ばれ、特に大きなものになるとタンパク質と呼ばれるようになります。これは一定の立体構造を持っていることが多いのですが、自然界に存在するタンパク質はアミノ酸配列とその立体構造がほぼ1対1に対応しているにもかかわらず(例外:アミロイドβとか一定条件下で二次構造を変化させるタンパク質とか)、現代の科学でも、アミノ酸配列だけから精度の良いタンパク質立体構造を予測することは難題とされています。これを解くための研究が世界中で行われており、その手法を競うコンテストCASPも開かれています。
テンプレートなしのタンパク質構造予測
構造がすでにProtein Data Bank(PDB)に登録されているタンパク質をテンプレート(鋳型)として、与えられたアミノ酸配列から構造を予想する方法はすでに様々なものが開発されています。その中でも最も使い勝手が良いなあと個人的に思うのがHHPredで、数日前の記事で紹介しました。しかし、与えられたアミノ酸配列が作るタンパク質の正解のフォールド構造が、これまでにPDBに登録されていないものであっても、(パーツごとにある程度類縁配列がたくさんあれば)これから紹介する手法であれば、かなりの精度で正解に近い構造を提示してくれると思われます。
余談
これを書いている中で12月3日にCASP13の結果が発表されて、DeepMind社の発表した"AlphaFold"が大差をつけて圧勝していましたね……。今回紹介する手法はこのランキングの中のBAKERに当たるものとほぼ同じだと思われます。また、図中のRaptorX系ってのは、以前紹介したCCMPredによる共進化のコンタクトマップ生成をDeep Learningで強化し、精度を大きく上げたものです。CASP12コンペでは、RaptorXが一定の範囲内の構造予測部門で1位のF1スコアを出していたそうです。(論文: https://doi.org/10.1371/journal.pcbi.1005324 )。
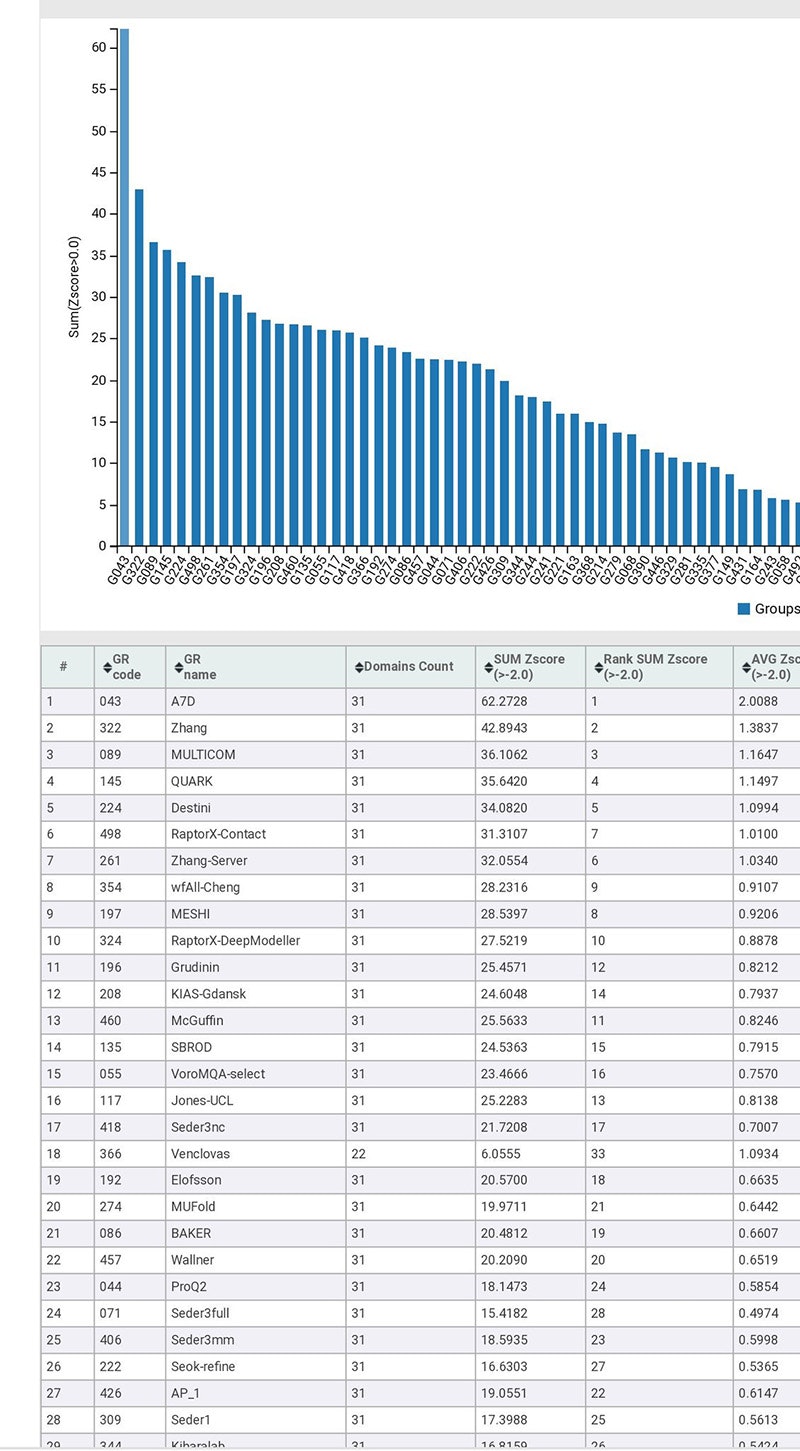
それでは、上記論文の内容を紹介します。
当該論文の簡単な内容紹介
Abstract
まとめると
- メタゲノムシーケンスデータのコンティグ によって、タンパク質をコードするアミノ酸配列のデータベースであるUniref100の配列空間を増強することで、CCMPredが作り出すタンパク質のコンタクトマップの精度を大幅に引き上げることができた。
- このコンタクトマップを距離拘束として加えながら、タンパク質モデリングソフトウェアRosettaの
AbinitioRelaxによるde novo構造モデリングを行った。 - まだ構造が1種類も報告されていない、614のタンパク質ファミリーに対してモデル構造の生成を行った。このうち206は膜タンパク質であり、137はProtein Data Bankにまだ存在しないフォールドを持つファミリーである。
- この計算手法は、Protein Structure Initiativeが原初の目的として思い描いていた、大きなタンパク質のファミリーの代表構造を提供するという野望を、わずかな労力だけで達成できることにつながるだろう。
本文の内容ダイジェスト
タンパク質ドメインのデータベースであるPfamには、(論文の公開時点で)14849のタンパク質ファミリーが登録されている。このうち4752ファミリーは最低でも1つの構造が実験的に結晶かまたはNMRによって決定されており、またさらに3984ファミリーはHHsearch fold-recognition programによって構造既知のホモログから比較モデリング法によって信頼性のあるモデルを生成することが可能である。残るは6113ファミリーであるが、902は信頼度のやや低いモデリングならまあできる程度、残りの5211ファミリーは構造情報がまったくないものである。
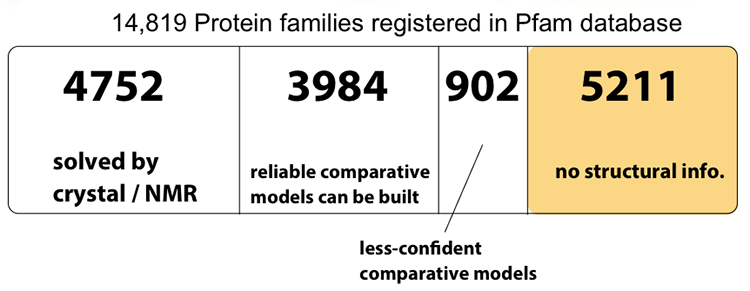
最近になるまでこの5211ファミリーについての精度の良いモデルを生成することはできなかった。Protein Structure Initiativeの当初の目的は、こうしたファミリーのうち最低でも1つの構造を決定することであったが、これは非常に難しい問題であるとわかり、このInitiativeの焦点は徐々に直近の生物学的な興味の方へとシフトしていった。
共進化情報を利用したコンタクトマップの生成とRosettaのde novo 構造予測を組み合わせた手法はすでに近年の58の大きなタンパク質ファミリーのモデル生成に使われている。cytochrome bd oxidaseの3つのサブユニットの例において(DOI:10.1126/science.aaf2477)、このcytochrome bd oxidaseの結晶データは得られたものの、位相情報は弱かったため、構造を決定づけることが難しい状態のままであった。この手法で作り出したサブユニット内&サブユニット間のコンタクトマップを利用した788残基の複合体のモデル構造は、3つのヘム鉄の位置と1つのメチオニンの位置と合わせて、結晶構造の位相を決定するのに使われ、最終的に登録された構造は我々が以前パブリッシュした大腸菌タンパク質のモデル構造にとても良く似ていた。CASP11構造予測実験では、複合体タンパク質構造を当てるコンテスト問題に対し、非常に正確なblind de novo予測が行えた。
CCMPredによるコンタクトマップ生成は、データベースUniref100の持つ配列量に依存する。正確な3Dモデル生成を導くのに十分なコンタクト部位予測精度を得るのに要求されるアライメント配列の数を決定するために、我々は構造既知である27の大きなタンパク質ファミリーについて、ベンチマークとしてのRosetta構造予測計算を行った。調べてみた結果、正確なモデル生成のためには、①ファミリー内の配列の数、②それら配列の多様性の数、そして③タンパク質の長さが大きく関係してくることに行き着いた。これら3つの要素を組み込んだ指標 $N_f$(: 80%配列同一性(sequence identity)クラスタリング閾値で得られた配列クラスタの数)÷(タンパク質長の平方根)) による測定はコンタクト部位予測での精度、および広い範囲でのファミリーに対し、モデル構造精度とよく相関していた。
現在までに構造がわかっていないタンパク質のファミリーについて、正確なモデル構造が作成できるために必要な$N_f$はどれくらいだろうか。Fig.1 に描かれているモデルはいずれも$N_f > 64$のファミリーについて生成されたものであり、それ未満の$N_f$では精度が落ちた。Fig 2Bに示すように、8%以下のファミリーでのみ$N_f \ge 64$だった。残る92%の構造未知のファミリーについて十分な精度で構造モデリングをすることは、Uniref100のデータベースから配列情報を使っていても当時はまだ不可能だった。
この構造モデリングの限界は、メタゲノムシーケンシングプロジェクトの研究分野によって大きく克服されることとなった。 これは複雑な生物学的サンプルに対してショットガンシーケンシングをかけるものだが、メタゲノム配列プロジェクトで決定されたタンパク質配列の数は、UniRef100データベースの増えていく配列の数よりかなり大きくなっている(Fig 2B)。メタゲノム配列データを含むことで、あるファミリーにおいては最大100倍ほど配列の数を増やすことができた。それにより、構造未知で、共進化情報を使う構造予測プログラムによって精度良く予測できるファミリーの割合は劇的に良くなった。$N_f \ge 64$となる割合のファミリーは8%から25%に増え、$N_f \ge 32$(のうち、良いフォールド精度を提示できたもの)では16%から33%まで増加した。
こうして得られたモデル構造のうち、構造未知のタンパク質フォールドを持つ中で重要な例として、developmental regulatorの1つであるChordin(骨形成タンパク質アンタゴニスト)、コバラミン合成に重要な酵素、メタロエンドペプチダーゼ、水銀・鉄トランスポーターなどがある。また、制限酵素XhoIもこの中に含まれる。ちなみに、この論文の原稿を執筆している間に、新規フォールドを持つタンパク質の構造が5種類ほど報告されたが、対応する我々のモデル構造とはTM-align score ≥ 0.7で類似していた(Fig S3 and Table S4)。
この論文で提示したモデル構造は全既知タンパク質ファミリーのうち約12%の構造情報を網羅するに至った。これが計算科学モデリングで達成されうるということはわずか5年前ではまったくもって予想できないことであった。ちなみに、なおも存在するこのモデリング手法の限界点の原因は、ほとんどの配列データが原核生物由来であり、そっち方面のデータが特に多いことに由来するものもあるが、今は菌類や真核生物のゲノムの構造予測シーケンシングプロジェクトが増えつつあるので、この手法はこうした真核生物の特定のタンパク質ファミリーにも適応できるようになっていくだろう。
結局どうやったらアミノ酸配列からモデル構造が作れるの?
やっていることは次の図の流れです。
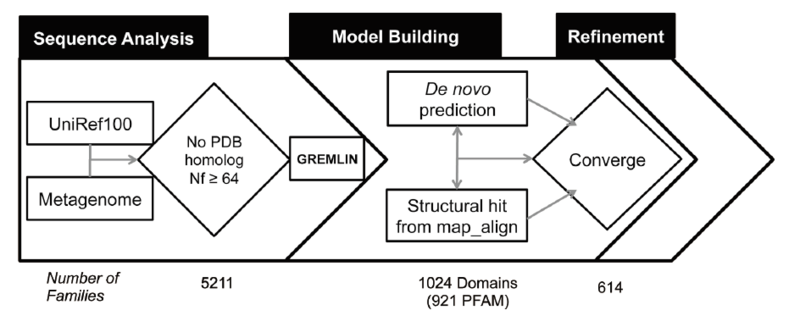
- 解析したいアミノ酸配列のFASTAファイルを用意する
- Uniref100(とメタゲノムシーケンスデータ)の配列データベースを使って、GREMLIN(またはそれを並列実装して高速化させたCCMPred)によるコンタクトマップ生成を行う(およそ30〜60分程度)
- 上記のコンタクトマップを拘束条件として取り入れながら、RosettaによるAbinitioモデリングを行う。残基数が長いほど計算時間は長くなるが、300残基ならば2〜3日程度で構造が収束する。
これだけです。
以上の内容を踏まえて、タンパク質構造を実際にモデリングするところまでやってみます。
マシン環境
以下の環境を想定しています。
- macOS High Sierra以降またはLinux OS, CentOS 7.4以降
- Rosetta 3.9以降がインストールされている
- CCMPredを用いたコンタクトマップ予測手法をマスターしている
まずはこの環境構築をがんばってください。
実例
いろいろファイルを使うことになるので、ここで紹介していくファイルのサンプルをGitHubに置いておきます。 https://github.com/BILAB/denovoprediction
以下のアミノ酸配列query.faについてのコンタクトマップ生成とRosettaのAbinitioモデリングをやってみます。
>WP_003963279.1_20
MTISVPQLDCPLSRPVHPEGERADAYAVEWLRGVGLMADEADAAPVLAVGLGRLAACYVDENASWDTLAF
MTILMAWYAEYDDRAIDSTGAIDGLTDAEVAELHRALGEILRDRPAPDPSDPVQRGLADVWRTLNGLASD
WDRAAFVDTTLRYFEANRYERVNIRRGIPPTPSAHIGMRRHGGHVYGMYILGAAVNGYRPERRVLDHAAV
RELETLAANYTSWANDLHSFAREHRMGQVNNLVWSVHHHEGLTFQQAADRVADLCDKELAAYLELRQTLP
ELGIPLTGATGRHVRFLEDMMWSMVDWSARSARYDVVPEAA
前回の記事にしたがって、CCMPredを使ったコンタクトマップ生成を行ってみると、次のようなhmmrenumbered.mapファイル(コンタクトマップ予想結果)が得られました。
CON 253 256 5.083 1.000
CON 73 133 4.321 1.000
CON 276 298 4.158 1.000
CON 180 219 4.141 1.000
CON 171 174 3.807 1.000
CON 255 259 3.454 1.000
CON 149 190 3.350 1.000
CON 250 260 3.094 1.000
(以下省略)
入力したquery.faのアミノ酸番号に対応しており、1行目は253番目と256番目のアミノ酸が、Score 5.083, Probability 1.00でコンタクトするだろうと予想されています。
これを、前回の記事で紹介したPyMOLのcontactmap表示プラグインで表示させてみます。載せる構造はHHPredで作った構造で作り出したもの( WP003963temp.pdb )を仮に表示します(本来このアミノ酸配列が作るタンパク質は構造未知なのですが、とりあえずここではホモロジーモデリングしたものを仮の正解として扱います)
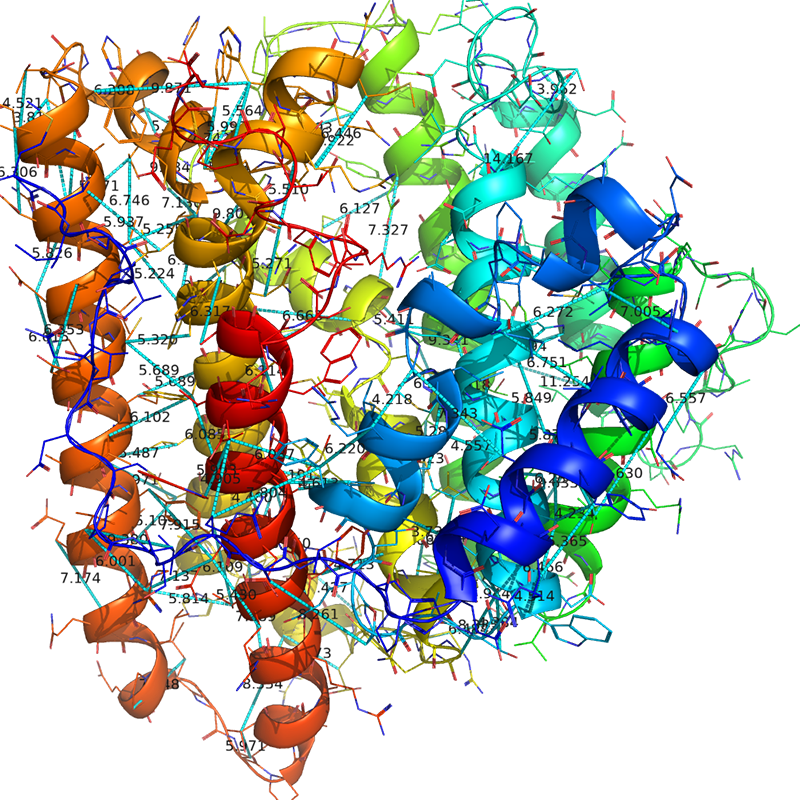
ホモロジーモデリングした構造の上でコンタクトマップの結果を見てみると、確かにだいたい予想された2つの残基ペアが近くなっているということがわかります。しかし、Scoreが小さくなるにつれて、明らかに「そうはならんやろ」というようなおかしい残基ペアがいくつか現れ始めます。……なので、Probability 0.999までのものを利用するか、上位n = (残基数L)/2 個だけの距離拘束を利用する、などする必要があります。
理論的には、完全なコンタクトマップが得られるのであれば、アミノ酸配列からタンパク質構造予測することはとても簡単になるわけなので、CCMPredをベースとしてDeep Learningをモリモリ使ってさらにコンタクトマップの精度を上げたものがいろいろと発表されています。RaptorXとか、DeepMindの発表した手法もこれがベースになっているはずです。
さて、本題に戻りまして、このファイルがある状態で、https://github.com/BILAB/scripts/blob/master/makecst.py にある変換スクリプト(@omi_UT 氏作成)でRosettaにおける距離拘束ファイルに変換します。
python3.6 makecst.py -f query.fa -m hmmrenumbered.map
変換後はこんな感じのfull.cst
AtomPair CB 253 CB 256 SCALARWEIGHTEDFUNC 5.083 SIGMOID 8.055 2.646
AtomPair CB 73 CB 133 SCALARWEIGHTEDFUNC 4.321 SIGMOID 7.442 3.861
AtomPair CB 276 CB 298 SCALARWEIGHTEDFUNC 4.158 SIGMOID 10.713 2.755
AtomPair CB 180 CB 219 SCALARWEIGHTEDFUNC 4.141 SIGMOID 10.135 2.688
(以下省略)
と、coarse.cst
AtomPair CB 253 CB 256 SCALARWEIGHTEDFUNC 5.083 BOUNDED 0 8.055 1 0.5 #
AtomPair CB 73 CB 133 SCALARWEIGHTEDFUNC 4.321 BOUNDED 0 7.442 1 0.5 #
AtomPair CB 276 CB 298 SCALARWEIGHTEDFUNC 4.158 BOUNDED 0 10.713 1 0.5 #
AtomPair CB 180 CB 219 SCALARWEIGHTEDFUNC 4.141 BOUNDED 0 10.135 1 0.5 #
(以下省略)
が作成されます。まずは上記アウトプットの
AtomPair CB 253 CB 256 SCALARWEIGHTEDFUNC 5.083 SIGMOID 8.055 2.646
は、これはRosettaの解説ページによれば、253番目のCβ原子と256番目のCβ原子の間に
f(x) = 5.083 \left( \frac{1}{1+\exp(-2.646(x-8.055))} - 0.5 \right)
という形でのシグモイド型距離拘束をかけています。2点間の平衡距離(ここでは8.055)は、該当する2つのアミノ酸の組み合わせで決めうちされているようです。同様に、
AtomPair CB 253 CB 256 SCALARWEIGHTEDFUNC 5.083 BOUNDED 0 8.055 1 0.5 #
の意味は……数式に表すのがめんどいので該当部分の箇所を読んでください(適当)
続くRosettaを使ったモデリングを行うときに、最初にcoarse.cstの方を用いて主鎖+Cβレベルの構造を作り出し、続いてそこから側鎖を生やして、full.cst型の距離拘束を用いながら構造を作り出すという流れになっています。
RosettaのAbinitio protein foldingで構造を作り出す
Rosetta Abinitio protein foldingとは
タンパク質モデリングソフトウェアRosettaのチュートリアルサイト(こことかこことか、後者のがメイン)に、だいたいの流れが書いてあります。
簡単に言えば、これまででもRosettaのAbinitio protein folding法は配列情報からのタンパク質モデリング法に定評があったのですが、上記のコンタクトマップの距離拘束を入れることで飛躍的に構造収束率が上がったということです。
Rosettaはモンテカルロシミュレーション的なノリで構造をたくさん作り出します(とても雑な表現)。そして、「構造が収束する」というのは、下の図の左側のように、作り出した無数の構造の、正解構造からのRMSD値と構造のエネルギー値の2次元プロットが、funnel型になっていくようになればOKという感じです。
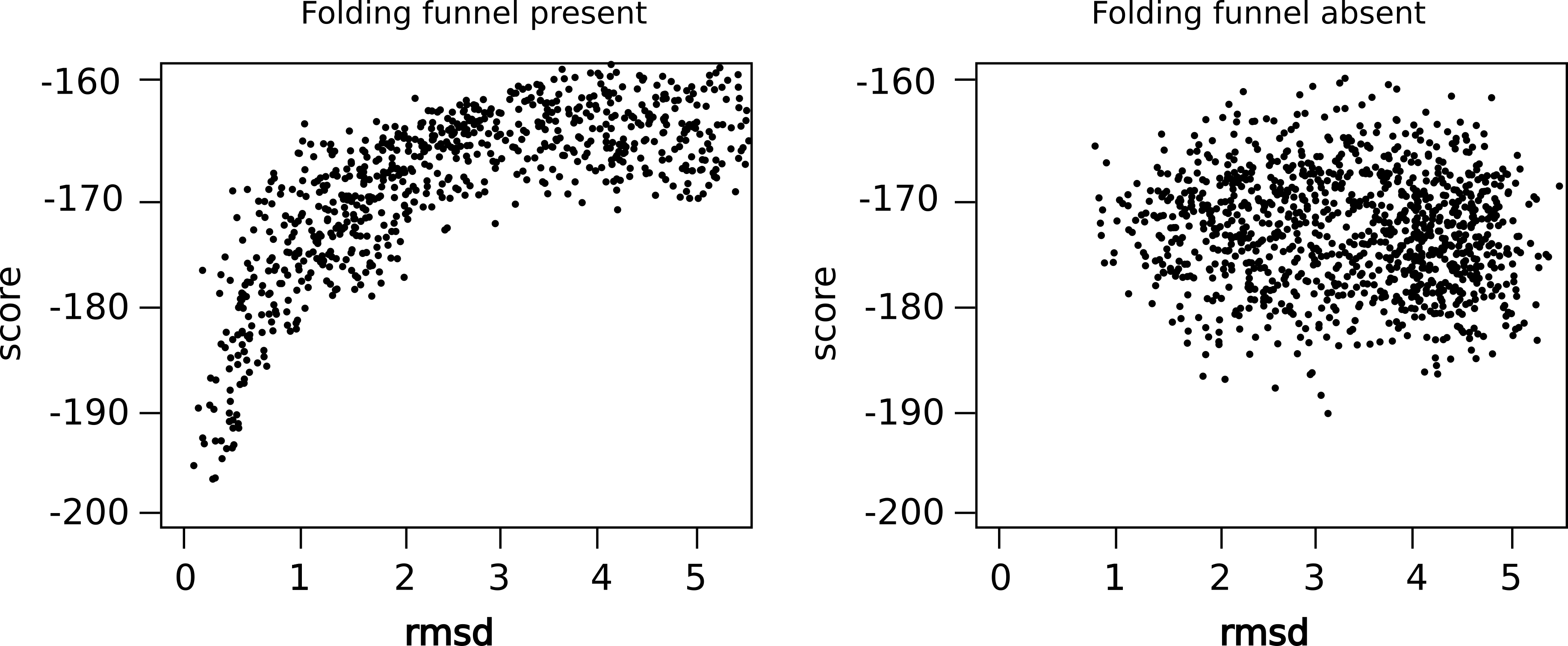
ただ、今回は正解構造はわかっていないので、この手法でモデルをたくさん作り出して一番小さいエネルギーを持つ構造を取り出すということになります。
実例Step 1. Fragment picking
まず、RosettaのAbinitio foldingを始めるにあたって、PSIPREDによる二次構造予測とそれに続くFragment Pickingの
予備動作を行います。PSIPREDは私が自前サーバー上に構築したVer 4.0のものを使って計算しました。でもWebサーバー版のもの(Ver 3.3)でもいいと思います。結果ファイルをWP_003963279.ss2とします。
続いて、RosettaのFragment Pickingを行います。Fragment pickingとは、クエリの配列情報やら二次構造予測情報を見ながら、それにマッチしていそうなPDBの構造フラグメントを拾ってくる処理のことです。ここでピックアップされたPDBの構造フラグメントをパーツとして使って、このあとのRosteta Abinitio protein foldingを実行します。なので、この段階のパラメータをいろいろいじることでどのパーツを拾ってくるかで、このあとのモデリング結果にも結構影響が出るので、選び方はいろいろ試して見る必要があります。が、ここではとりあえずsimple.wghtsというファイルに
# score name priority wght max_allowed extras
RamaScore 400 2.0 - predA
SecondarySimilarity 350 1.5 - predA
FragmentCrmsd 0 0.0 -
という内容でフラグメントの選び方のパラメータを書いておきます。これを用意した状態で、flagsファイルを作成します。
# Input databases
-in::file::vall /path/to/Rosetta3.10/tools/fragment_tools/vall.jul19.2011.gz
# Query-related input files
-in::file::fasta query.fa
-in::file::s WP003963temp.pdb
-frags::ss_pred WP_003963279.ss2 predA
# Weights file
-frags::scoring::config simple.wghts
# What should we do?
-frags::bounded_protocol
-out::file::frag_prefix frags
-frags::describe_fragments frags.fsc
ここで、-in::file::vallにはお使いのマシン環境にインストールされているRosettaのPATHを入れ、tools/fragment_tools/vall.jul19.2011.gzまでのPATHを完成させてください。また、query.fa, WP003963temp.pdb, WP_003963279.ss2, simple.wghtsの4ファイルを同じディレクトリに入れておきます。
これを置いたら、
/path/to/Rosetta3.10/main/source/bin/fragment_picker @flags
を実行して、Fragment picking処理を実行します。しばらくすると、frags.200.3mers, frags.200.9mers, frags.fsc.200.3mers, frags.fsc.200.9mersの4ファイルが作成されます。
実例Step 2. Ab initio Protein Folding
Fragment Pickingが終わったら、Ab initio Protein Foldingに移ります。
まずoptionsというファイルを作成します。
# Make sure all variable names have been replaced with absolute path and that no line begins with a $ or ~s
-in
-file
-native WP003963temp.pdb # native PDB file (optional)
-fasta query.fa # protein sequence in fasta format
-frag3 frags.200.3mers # protein 3-residue fragments file
-frag9 frags.200.9mers # protein 9-residue fragments file
-constraints
-cst_weight 1.5
-cst_file coarse.cst
-cst_fa_weight 1.5
-cst_fa_file full.cst
-abinitio
-relax
-increase_cycles 10 # Increase the number of cycles at each stage in AbinitioRelax by this factor
-rg_reweight 0.5 # Reweight contribution of radius of gyration to total score by this scale factor
-rsd_wt_helix 1.0 # Reweight env, pair, and cb scores for helix residues by this factor
-rsd_wt_loop 0.5 # Reweight env, pair, and cb scores for loop residues by this factor
-relax
-fast # At the end of the de novo protein_folding, do a relax step of type "FastRelax". This has been shown to be the best deal for speed and robustness.
-out
-nstruct 2000 # how many structures do you want to generate? Usually want to fold at least 1,000.
-file
-scorefile score.sc
-silent silent.out
-overwrite # overwrite any existing output with the same name you may have generated
-nstruct 2000
2回出てくる-nstructのところには2回同じ値を入れる必要があるみたいです(?) 最低でも2000くらいは設定するほうが良いでしょう。
これを作成したら、mpirun -np 12 /path/to/Rosetta3.10/main/source/bin/AbinitioRelax.mpi.linuxgccrelease @optionsを実行します。これはOpenMPIで並列化しており、CPU数は -npのところに入力しています。並列化しないと1コアしか使ってくれないので、並列化版を使うことを強く勧めます。
計算が始まってしばらくすると、silent_n.outのファイルとかscore.fscのファイルが生成されます。この.outの方には構造情報が圧縮されて出てきています。また、作成したモデルのスコア値がscore.fscに格納されます。ここは数日ほど時間がかかるのでじっと待ちましょう。途中でも各ファイルを開くことは可能です。
実例Step 3. モデル構造の取り出し
計算が始まってしばらく経ったら、それまでに作り出されたタンパク質構造のエネルギースコアをソートして見て見るために、sort -k2 score.fsc | less -SNのコマンドを入れてみましょう。これは、score.fscの2番目のカラム(エネルギースコア)をソートして、さらにlessコマンドのオプションで右端折返しをしないまま表示してくれます。
1500個くらいやった結果はこんな感じで表示されました。(もっと個数を増やして計算した方がより良いと思います)
SCORE: score fa_atr fa_rep fa_sol (中略) description
(一番下の行までスキップ)
SCORE: -811.198 -1860.934 269.154 1201.832 (中略) S_00000887
SCORE: -812.113 -1870.602 275.892 1214.503 (中略) S_00001009
SCORE: -824.378 -1897.790 294.309 1240.949 (中略) S_00000467
これは、計算が始まってS_00000467のIDを持つ構造がそれまで計算されたモデル構造の中で最も低いスコアを持っていることを表しています。この構造を次のコマンドで取り出します。
extract_pdbs -in:file:silent silent_*.out -tags S_00000467
こうして取り出してみたタンパク質構造S_00000467.pdb(カラー)をHHPredの結果(白色)と比較してみます。
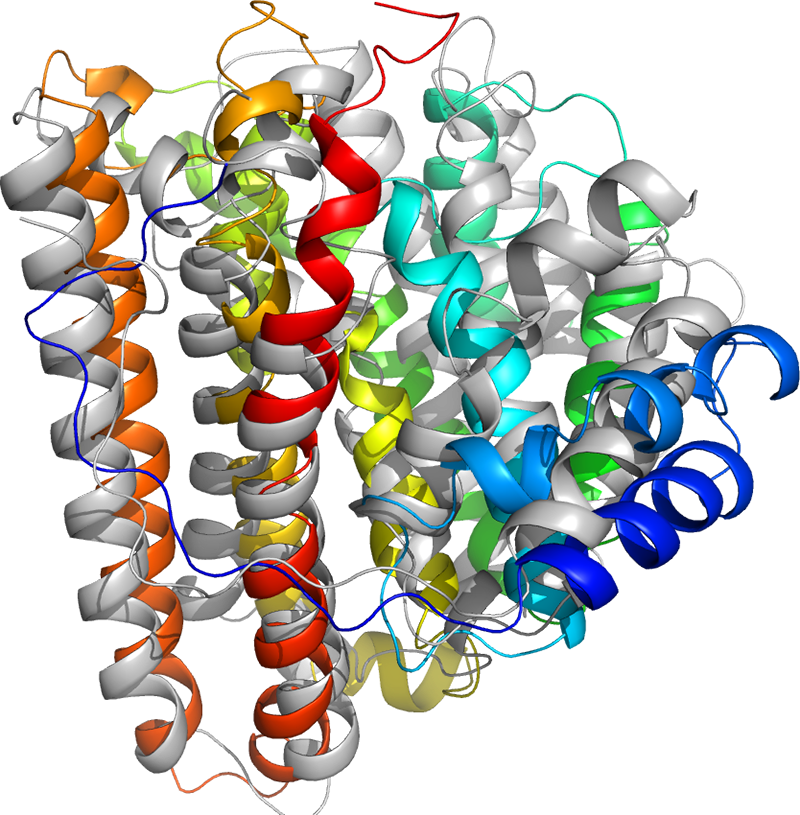
RMSDは5.2Å程度でした。タンパク質の概形としてはうまくモデリングできていますし、フォールドの方向性も大きな間違いな部分はありませんでした。ただ、ちょいちょいモデリングが怪しい部分があったり、HHPredと一致していない部分が見えますね……。
このAbinitio法で作り出したモデルとHHPredのモデルとで、どっちが正解なのかはわかりませんが、まあぼちぼちうまく行ったんじゃないでしょうか。
気になる方は、任意の構造既知のタンパク質でこの手法を使ってみて、実際に構造が一致するかどうか試してみましょう。
Pros/Cons
Pros
論文にある通り、PDBに1つも類似構造がないものでもモデル構造をうまく作成することができます。また、CCMPredの精度が類縁配列の本数に依存しているため、似たようなタンパク質が多くある系のモデリングではうまくいきやすいです。論文では膜タンパク質のモデリングも多く報告されていました(てかむしろ膜タンパク質の方がうまく行きやすい?)。
Cons
計算するのにそこそこ時間がかかります。タンパク質の全体構造についての精度は結構良いが、逆に基質結合部位・機能部位周辺のモデリングについてはそんなに精度が良くない(むしろ悪い)。基質結合部位とか活性部位については、HHPredが利用できるのであればそちらを利用したほうが圧倒的にきれいにモデリングできます。なので足場タンパク質とかには良いのかもしれないです。HHPredや他の手法とうまく組み合わせるとよいのかもしれないですね。
精度を上げるために
(書きかけ)
- より良いコンタクトマップ生成法を利用する (RaptorXとか)
- RosettaのFragment Pickingの段階で様々なパラメータを試してみる
- DeepMindがやったように、角度・二面角の拘束を入れてみる(手法は謎ですが)
おわりに
この記事は創薬Advent Calendar 2018の記事として書いたわけですが、まあタンパク質の形状がわかったからと言って、それですぐ創薬に結びつくかと言われると、そうではないと思います。実際にはタンパク質の形状が完全に判明していても、それに結合する化合物をデザインする**Structure-based drug design(SBDD)**の方法(これについては1年前の記事を読んでください)は、現代ではまだLigand-basedの方法に比べてヒット率が良くないという傾向があり、ここの改善については他の方面からのブレイクスルーが待たれるところではあります。これを読んでくれている製薬会社の人は期待させてすまんやで。
ですが、SBDDの創薬ならではの強みもあり、まだまだ構造が解かれていない生活に重要なタンパク質というのはたくさんあるので、このようにしてタンパク質モデル構造を提示できるというのは、創薬の研究者だけでなく、多くの生命科学の研究者にとって役立つものなのではないかなと思います。