Introduction
タンパク質のコンタクトマップ とは、タンパク質が折り畳まれていったときに、どのアミノ酸残基とどのアミノ酸とが近くなったかという距離の情報を2次元マップで表したものです。例として、157残基長のPDB: 5DIRのChain Aのコンタクトマップは次の白黒画像のようになります。


この記事では、なんと、アミノ酸配列だけから、このコンタクトマップの予想を高精度で返してくれる プログラムである GREMLIN/CCMPred の使い方を紹介します。
GREMLINは、David Baker研究室から2013年に発表された、共進化情報を利用した強力なコンタクトマップ予測法 であり(DOI: 10.1073/pnas.1314045110)、CCMPredはGREMLINを並列化して大幅に高速化したプログラムとなっています(DOI: 10.1103/PhysRevE.87.012707 , DOI: 10.1002/prot.22934)。
GREMLIN/CCMPredは**Direct coupling analysis (DCA)**の一種として分類されることもあり、類似のウェブサービスはEVcouplingsやライス大学のDCA Webserviceなどがあります。
はじめに、ウェブサーバー版GREMLINを使った結果を紹介しながら、それを自前のパソコン・サーバー上で動かす方法を紹介していきます。
何に使えるの?
正確なコンタクトマップが得られるのならば、配列からのタンパク質構造予測に強力に役立ちます。
計算環境準備
マシン環境
- macOS 10.13.6 (High Sierra), 2.8GHz Intel Core i7, メモリ16GB またはLinux OS
- macOSの場合ターミナルからHomebrewをインストールしてある
- 100GB以上のディスク容量を持つストレージ。SSDであればさらに動作が早い。
- 類縁配列検索ソフトウェアHH-Suiteのインストールをしてある
配列検索のソフトウェアHMMERのインストール
macOSの場合
Homebrewで簡単にインストールできる。 brew install hmmer で終了
Linux OSの場合
MPIを用いた並列計算をしたい場合は./configure時に --enable-mpi を付けるべき。 --prefix でインストール先を指定する。今回の例では/usr/local/package/hmmer/3.2.1 にしておきます。
wget http://eddylab.org/software/hmmer/hmmer-3.2.1.tar.gz
tar zxvf hmmer-3.2.1 && cd hmmer-3.2.1
./configure --prefix=/usr/local/package/hmmer/3.2.1 --enable-sse --enable-threads
make -j8
make -j8 install
# miniappsの追加インストール
cd easel
make install
map_alignのインストール
map_alignは2つのコンタクトマップが得られたときに、それらの中でオーバーラップしている領域を最大化し、ギャップを最小化するための簡単なプログラムです。あとでちょっと必要になるので、こいつもインストールしておきます。GitHubは https://github.com/sokrypton/map_align
map_align takes two contact maps and returns an alignment that attempts to maximize the number of overlapping contacts while minimizing the number of gaps.
インストール方法は
mkdir -p /path/to/apps/map_align #インストール先ディレクトリを適当に作る
cd /path/to/apps
git clone https://github.com/sokrypton/map_align.git
cd map_align
g++ -O3 -std=c++0x -o map_align main.cpp # map_alignプログラムのコンパイル
# binディレクトリを作り、その中にプログラムを入れておく
mkdir bin
cp mk_map/*.pl ./bin
cp map_align ./bin
# 実行権限を付与(permission denied対策)
chmod +x ./bin/*
上記の処理後、export PATH="/path/to/apps/map_align/bin:$PATH"として、ここにPATHを通しておきます。
CCMPredのインストール
GREMLINの並列&GPU計算実装版のCCMpred ver.0.3.2をインストールします(現行は0.3.3 alpha)。NVIDIA GPU/CUDAによる高速化に対応しており、インストール時にGPU対応オプションを指定しておくと5倍速くらいになりますが、CPUだけでも数分の処理時間なので、まあなくてもどうにかなります。
macOS, LinuxOSの場合も、いずれもCMakeをインストールしておく必要があります。CMakeのバージョンは3.14以上の最新版を使っている方が無難です。Ubuntu 18.04にaptでインストールできるCMakeはver.3.10.2ですが、nvcc fatal : Unknown option 'fopenmp'と出てしまいインストールに失敗します。macOSならば brew install cmake でインストールできます。
インストール方法は
# gitでダウンロード
git clone --recursive https://github.com/soedinglab/CCMpred.git
cd CCMpred
mkdir build ; cd build
# GPU/CUDAを使いたい場合は指定……しなくても勝手に検知してくれることがある。
# -DCUDA_TOOLKIT_ROOT_DIR= でCUDAドライバの場所を指定。
# CUDAを使わない場合は-DWITH_CUDA=OFFをcmake時のオプションに追加する
# ここでインストール先は/usr/local/package/CCMpred/0.3.2とする(任意)
cmake .. -DCMAKE_C_COMPILER=/bin/gcc -DCMAKE_CXX_COMPILER=/bin/g++ -DCMAKE_INSTALL_PREFIX=/usr/local/package/CCMpred/0.3.2 -DCUDA_TOOLKIT_ROOT_DIR=/usr/local/cuda
make -j4
make install
これでインストール完了。簡単な使い方は次のような感じ
$ ccmpred -h
Usage: ccmpred [options] input.aln output.mat
Options:
-d DEVICE Calculate on CUDA device number DEVICE (set to -1 to use CPU) [default: 0]
-t THREADS Calculate using THREADS threads on the CPU (automatically disables CUDA if available) [default: 1]
-n NUMITER Compute a maximum of NUMITER operations [default: 50]
-e EPSILON Set convergence criterion for minimum decrease in the last K iterations to EPSILON [default: 0.01]
-k LASTK Set K parameter for convergence criterion to LASTK [default: 5]
-i INIFILE Read initial weights from INIFILE
-r RAWFILE Store raw prediction matrix in RAWFILE
-w IDTHRES Set sequence reweighting identity threshold to IDTHRES [default: 0.8]
-l LFACTOR Set pairwise regularization coefficients to LFACTOR * (L-1) [default: 0.2]
-A Disable average product correction (APC)
-R Re-normalize output matrix to [0,1]
-h Display help
ヘルプを読んだら実行。-tにはCPU数、-nにMax Iteration回数、-dでGPUのナンバー(通常は0)を指定する。その後インプット・アウトプットファイル名の指定。
HH-suiteのインストール
上の 類縁配列検索ソフトウェアHH-Suiteのインストールを参考に、プログラムをインストールしておきます。
データベースの構築
Uniclust30データベースと、Uniref100データベースの2つのセットアップが必要です。
uniclust30_2018_08データベースの用意
類縁配列検索ソフトウェアHH-Suiteのインストールの中で紹介してありますので、そちらを参照してください。
Uniref100データベースの用意
ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref100 からダウンロードし、BLAST+のプログラムmakeblastdbでデータベース構築をします。BLAST+のインストール方法はここでは割愛します。
mkdir -p /path/to/database/uniref100 && cd /path/to/database/uniref100
# uniref100は34.6 GB以上のファイルサイズ
wget ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref100/uniref100.fasta.gz
gunzip uniref100.fasta.gz
makeblastdb -title uniref100 -dbtype prot -in uniref100.fasta -max_file_sz 2GB
GREMLIN webサーバー版を使ってコンタクトマップ予測をしてみる
Baker研究室がGREMLINのwebサーバー版を公開してくれています。ここに投げたクエリ配列とその結果をもとに、自前マシンでの再現実験のときの指標にします。
http://gremlin.bakerlab.org/submit.php に向けて配列を投げてみます。テストケースとして、膜タンパク質PDB: 5DIRのChain Aについてやってみます。これは、当時Pfamで新規ドメインとして認識され、類似構造が存在していないタンパク質構造とされていたものです。なので、この構造が報告されるまではHHPredなどのホモロジーモデリング手法ではうまくいかなかったそうです。
>querytest(5DIR_A)
LVPRGSHMPDVDRFGRLPWLWITVLVFVLDQVSKAFFQAELSMYQQIVVIPDLFSWTLAYNTGAAFSFLADSSGWQRWLF
ALIAIVVSASLVVWLKRLKKGETWLAIALALVLGGALGNLYDRMVLGHVVDFILVHWQNRWYFPAFNLADSAITVGAVML
ALDMF
(見る人が見たら、最初にLVPRGSっていうthrombin認識配列がついていることがわかります)これをGREMLINのフォームに入力しますが、オプション設定は以下の画像のように設定しました。
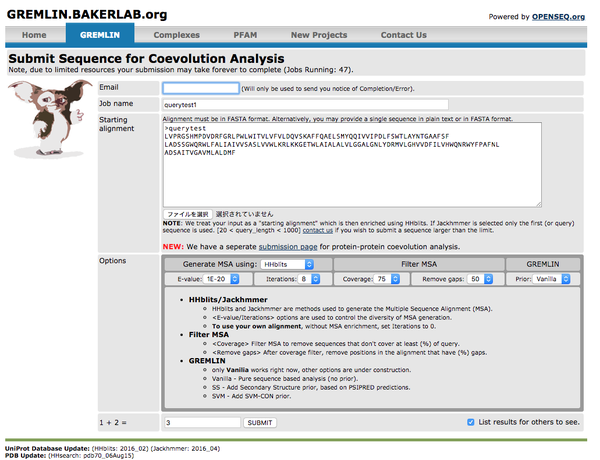
Generate MSA using:HHblitsにし、E-value:は1E-20、Iteration:は8にした。Filter
MSAについてはCoverage: 75、Remove gaps:50とした。
10日ほど待つと(このときはジョブ待ちが非常に長かった)、結果はこんな感じで返ってきました。
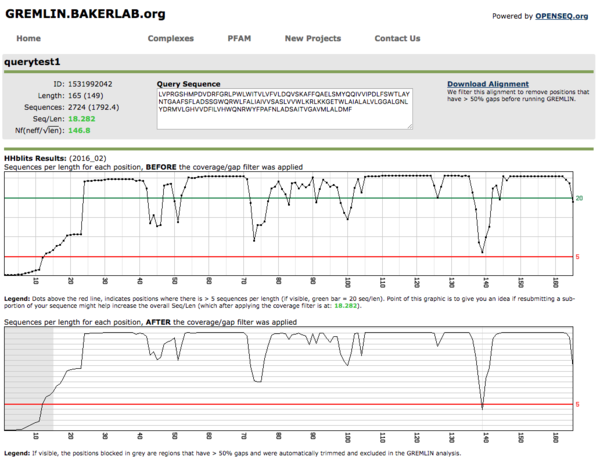
右上のDownload alignmentにはフィルタリングされる前のアライン結果が表示されます。Seq/Lenというのは、hhblitsで得られたMSAの配列数がSeq, クエリの配列長をLenとして割った値です。()内の数字はおそらく実効の値で、Lenでは最初の15アミノ酸くらいでMSA中で50%以上のギャップが存在していたため、その部分の配列は実効値にカウントされなかったと考えられる。Seqの実効値は上のCoverage: 75%フィルターで減ったのかな?ちょっと自信がないですが、多分そういうことだと思います(適当)。
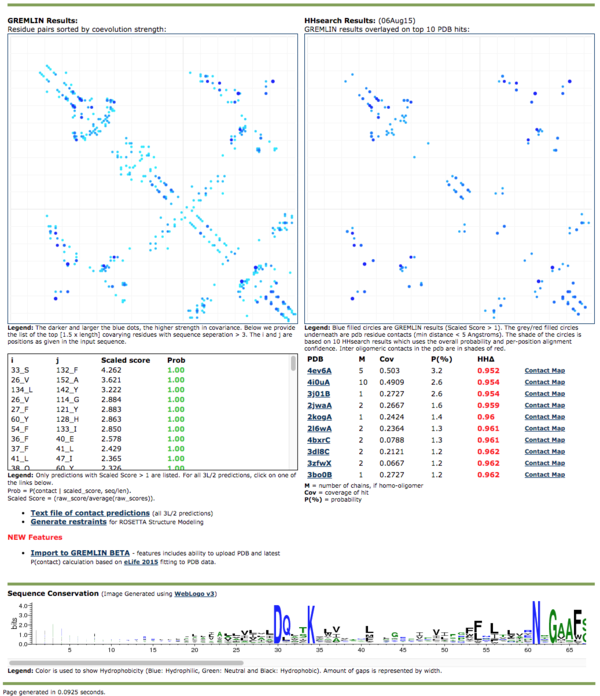
左にGREMLINが出したContact Mapが示されています。最初に示した白黒画像でのコンタクトマップとは違って、上下反転しているので気をつけて比較してください。
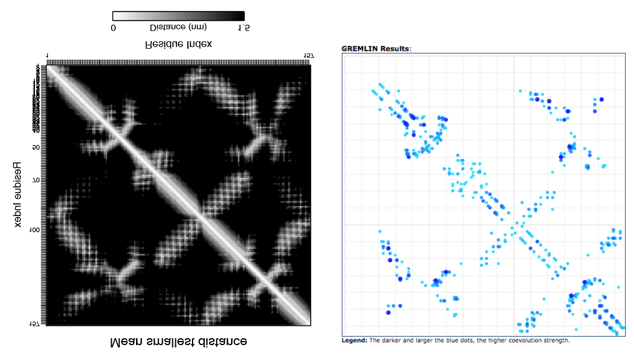
並べてみると、だいたいマッチしていることがわかりますね。
その下に共進化情報に基づいてコンタクトすると考えられる残基ペアのリストが書かれています。
下のNew Features!のところでImport to GREMLIN BETAを押すと、以下の画面が表示されました。
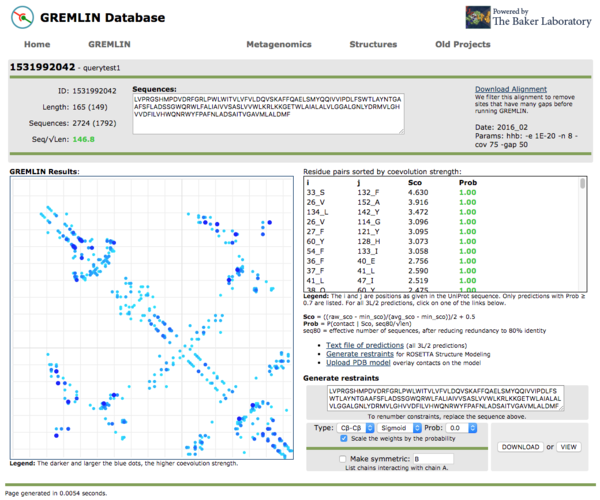
情報に大きく違いはないように見えますが……?
さて、まあこうした内容を、in houseで出力させるにはどうしたらいいかを考えてみます。
自前マシンでGREMLIN/CCMPredによるコンタクトマップ生成を行う
上のWebサーバーになげたときのクエリ配列を使って再現実験をしてみます。
まずはHH-suiteとHMMER, map_alignのコマンドが使えることを確認しておきます。
export PATH="/path/to/apps/map_align/bin:$PATH"
which map_align
which hhblits
which hmmbuild
echo $HHLIB
としてみて、PATHが通っていることと、環境変数HHLIBが存在することを確認します。上のセットアップ通りにやっていれば大丈夫なはずです。
まずhhblitsをクエリ配列が入っているファイルquery.faについて、以下のオプションとともに行います。
hhblits -cpu 12 -i query.fa \
-d /Users/Agsmith/database/hhsuite/uniclust30_2018_08/uniclust30_2018_08 \
-oa3m query.a3m -n 8 -e 1E-20 -neffmax 20 -nodiff -maxfilt 999999 \
-realign_max 999999
ここで-cpuはCPU数を指定します。使っているマシンの最大CPU数を指定すると良いでしょう。結果として得られるquery.a3mを上のGREMLINウェブサーバーの結果と比較してみたのですが、この時点ですでに10683本の類似配列が釣れてきました(ウェブサーバーは2724本)。-neffmax 20 -nodiff -maxfilt 999999 -realign_max 999999このへんのオプションが効いているんでしょうかね?
続いてこれをhhfilterでフィルタリングします。
hhfilter -i query.a3m -o query_filt.a3m -id 90 -cov 75
得られたこのファイルについて、reformat.plでa2mファイル形式に変換します。これは続くhmmbuildでHMMの生成を行うためです。
${HHLIB}/scripts/reformat.pl query_filt.a3m query_filt.a2m
hmmbuild --cpu 12 hmmout query_filt.a2m
これにより、作成されたHMMがhmmoutファイルに出力されます。
続くhmmsearchのステップでは、このhmmoutと対象データベース(ここではUniref100)をもとに、hmmsearch [options] <hmmfile> <seqdb>の形式で指定します。
hmmsearch --cpu 28 -A hmmsearch.sto --tblout tblout.txt --incT 27 \
hmmout /Users/Agsmith/database/blast/db/FASTA/uniref100/uniref100 > hmmsearch.out
--incT 27というのがPfam境界定義にも使われるという、bit-scoreのしきい値27の指定です。また、得られるMSAを-A hmmsearch.stoで出力させます。この出力形式はstockholmフォーマットになっていますため、続くフィルタリングではreformat.plでa2m形式に変換してから処理を行います。
得られたhmmsearch.stoに対し、再びフィルタリングをかけます。
${HHLIB}/scripts/reformat.pl hmmsearch.sto hmmsearch.a2m
hhfilter -i hmmsearch.a2m -o hmmsearch_filt.a3m -id 90 -cov 75
さて、これからCCMpredにかけてcontact mapを得たいわけなのですが、このhmmsearch_filt.a3mは現時点では
>UniRef100_A0A1W9WY37/34-184
SNANALIWLWLSLLVVVLDQVTKVWVSATLELHQSIAVLPFFDLRLVHNEGAAWSFLADASGWQRWFLSGLAILISGVIVVWLSRLERQQRWLACALALILGGALGNVMDRAMYGYVVDFIDIYYQQMHWPVFNIADSAISVGAVMLLIDA
>UniRef100_A0A148NB86/5-151
----HVKWLGLSVAVIVLDQLTKWWVVSTLRLHESIPVWPFFNLTYVRNTGAAFSFLAGAGGWQRWFFLGLAVVASMAILVWMWRLKDDEVLTAAGLSLVLGGALGNFIDRAAYGYVIDFLDFYIQNWHWPAFNVADSAITIGVILLLWDG
>UniRef100_UPI0009EEDF12/24-173
-VSRYRLLAIVTAVVLVLDQATKLYVDSHFRLHESVTVLEnFFHFTYVRNKGAAFGIFADS-AIRIPFFITVSLVAAVGILWYLRRLKDNQKLVCFSLALIFSGALGNLIDRVRFGEVIDFIDVHWYQYHWPAFNVADSAISVGVTLLLVDL
>UniRef100_A0A0A6P993/4-156
kSSEKALVWFWLSLLVVILDQVTKVWISANLALYQSIVVLPFFNLTLLHNEGAAWSILATAGGWQRWFLSGLAIVISGVLMIWLSRLKRQQRWLACALALILGGALGNVIDRVIYGYVVDFIDVYYNhEWHWPAFNIADSAISVGAVMLLIDA
>UniRef100_UPI00041D792D/6-151
-----RLLLIISAVVLVLDQATKLYIDSRFALHQSLTVVEnFFHITYVRNKGAAFGIFADS-AVRIPFFITVSIVAAIGIVWYLHRLNEQQRLLHVALSLIFAGAVGNLIDRIRLGEVIDFIDVHWYQYHWPAFNIADSAITVGVGILLLDL
こんな感じとなっており、FASTA形式でかつ配列長が揃っていません。CCMpredの計算処理をさせるためには、PSICOV式フォーマットになっていることと、配列長が揃っていること が条件だそうです(参考:https://github.com/soedinglab/CCMpred/wiki/FAQ )。
CCMpredのディレクトリの中にもscripts/ディレクトリの中にその問題を解決してくれるスクリプトがあるのですが、それよりも便利なものがmap_alignの中のmk_mapディレクトリに同梱されていました。ここの中にあるa3m2alncut.pl, mk_map.pl, pdb2map.plはこのcontact map作成に非常に便利なものです。
まずは https://github.com/sokrypton/map_align/blob/master/mk_map/cmd を参考にして、hmmsearch_filt.a3mからPSICOVインプットファイルを生成します。
input=hmmsearch_filt ; a3m2alncut.pl ${input}.a3m ${input}.aln ${input}.cut
このa3m2alncut.pl内部で50%以上のギャップ位置の削除と75%未満のcoverage削除を行ってくれています。こうして、CCMpredのインプットとなるhmmsearch_filt.aln(下のMSA)とhmmsearch_filt.cutが生成されます。
ALIWLWLSLLVVVLDQVTKVWVSATLELHQSIAVLPFFDLRLVHNEGAAWSFLADASGWQRWFLSGLAILISGVIVVWLSRLERQQRWLACALALILGGALGNVMDRAMYGYVVDFIDIYYQQMHWPVFNIADSAISVGAVMLLIDA
HVKWLGLSVAVIVLDQLTKWWVVSTLRLHESIPVWPFFNLTYVRNTGAAFSFLAGAGGWQRWFFLGLAVVASMAILVWMWRLKDDEVLTAAGLSLVLGGALGNFIDRAAYGYVIDFLDFYIQNWHWPAFNVADSAITIGVILLLWDG
YRLLAIVTAVVLVLDQATKLYVDSHFRLHESVTVLEFFHFTYVRNKGAAFGIFADS-AIRIPFFITVSLVAAVGILWYLRRLKDNQKLVCFSLALIFSGALGNLIDRVRFGEVIDFIDVHWYQYHWPAFNVADSAISVGVTLLLVDL
ALVWFWLSLLVVILDQVTKVWISANLALYQSIVVLPFFNLTLLHNEGAAWSILATAGGWQRWFLSGLAIVISGVLMIWLSRLKRQQRWLACALALILGGALGNVIDRVIYGYVVDFIDVYYNEWHWPAFNIADSAISVGAVMLLIDA
-RLLLIISAVVLVLDQATKLYIDSRFALHQSLTVVEFFHITYVRNKGAAFGIFADS-AVRIPFFITVSIVAAIGIVWYLHRLNEQQRLLHVALSLIFAGAVGNLIDRIRLGEVIDFIDVHWYQYHWPAFNIADSAITVGVGILLLDL
GLRWLWISFIVLLLDQATKWWASSALELYQPVPVLPYLNFTLLHNPGAAFSFLADAGGWQRWFFTGLAVVISAVIVMWLARLSPQQRMLGIALALVLGGAVGNVIDRFIYGYVIDFIDVYYQTWHWPAFNIADSAITIGAVLLLIDV
hmmsearch_filt.cutの方はあとでcontact mapを生成する時に使います。
さて、いよいよCCMpredを使った計算を行います。-t 12というのはCPU数を指定します。収束を確実にするために-n 100を付けておきます。
$ ccmpred -t 12 -n 100 hmmsearch_filt.aln hmmsearch_filt.mtx
_____ _____ _____ _
| | | |___ ___ ___ _| |
| --| --| | | | . | _| -_| . |
|_____|_____|_|_|_| _|_| |___|___|
|_|
using CPU (12 thread(s))
Reweighted 12998 sequences with threshold 0.8 to Beff=9296.36 weight mean=0.715215, min=0.0144928, max=1
Will optimize 9532509 32-bit variables
iter eval f(x) ║x║ ║g║ step
1 1 2.41303e+06 25531.6 8.7357614e+09 9.75e-06
2 1 2.36412e+06 25531 7.3350098e+09 6.19e-06
3 1 2.31537e+06 25531 6.0129638e+09 5.16e-06
4 1 2.26682e+06 25533.3 4.817707e+09 4.99e-06
5 1 2.21858e+06 25538.5 3.8217572e+09 5.31e-06
6 1 2.17071e+06 25549.4 3.0241239e+09 6.01e-06
7 1 2.1231e+06 25568 2.3991114e+09 7.08e-06
.....
55 1 1.33715e+06 41937.3 55085000 1.95e-05
56 1 1.3343e+06 41937.2 50249676 2.1e-05
57 1 1.33154e+06 41931.3 51348760 2.46e-05
58 1 1.32892e+06 41920.5 48426468 2.7e-05
59 1 1.32657e+06 41904.9 56314588 3.43e-05
Done with status code 0 - Success!
Final fx = 1325345.875000
xnorm = 93.2845
Output can be found in hmmsearch_filt.mtx
こんな感じで、2~5分くらいで返ってきました。GPU acceleratedにするとさらに5倍くらいは早くなりますが、このくらいの計算時間でしたらCPU onlyでも問題にならないくらいです(ただしCPU並列数指定オプションを忘れると悲惨)。hmmsearch_filt.mtxは例によって対称行列のようになっているはずです。ちなみに、ccmpredの-dオプションのところでNf計算(number of sequenses >80% identity)の80%しきい値を他の値に変えられるようです。デフォルトは-d 0.8となっているので、ここでは明示的に指定しませんでした。
このhmmsearch_filt.mtxをmk_map.plでmap形式に変換します。
input=hmmsearch_filt ; mk_map.pl -aln ${input}.aln -chk ${input}.chk -cut ${input}.cut -mtx ${input}.mtx -map ${input}.map -do_apc
得られたhmmsearch_filt.mapはこんな感じ
LEN 151
CON 14 138 1.000
CON 21 119 1.000
CON 47 115 1.000
CON 121 128 1.000
CON 14 101 1.000
CON 26 117 1.000
CON 15 108 1.000
CON 24 28 1.000
CON 41 120 1.000
CON 42 121 1.000
……
LEN 151の151は実効的な配列長です。また、この残基番号の数字は0-originになっていることに気をつけてください。query配列が165アミノ酸であり、50%ギャップの位置情報は失われているので、14アミノ酸分が消し飛んでつながっていることになります。つまり、これだけでは本来のクエリ配列長と照らし合わせることができません。しかし、最初の方で作成したquery_filt.a3mにはqueryの配列情報がまだ残っています。これを使って上の流れと同様にquery_filt.mtxとquery_filt.mapを作成します。このquery_filt.mapを見てみると
LEN 165
CON 32 131 1.000
CON 25 151 1.000
CON 133 141 1.000
CON 26 120 1.000
CON 59 127 1.000
CON 53 132 1.000
CON 25 113 1.000
CON 35 39 1.000
CON 37 59 1.000
CON 40 46 1.000
CON 36 40 1.000
……
こんな感じでした。これはGREMLIN ウェブサーバの結果と(0-originのために残基番号が1ずれていることを考慮すれば)同一となっています。
というわけで、2つのContact map:hmmsearch_filt.mapとquery_filt.mapが得られました。ここで、query_filt.mapにはquery配列の情報が完全に残っているので、この2つをContact mapをうまいことマッチングさせるのにmap_alignを使います。
map_align -a query_filt.map -b hmmsearch_filt.map > mapalignout.txt
-aと-bには2つのmapファイルを指定します(順不同)。結果は標準出力で返ってくるので、mapalignout.txtに入れておきます。これを見てみると、
MAX 0_1_0 query_filt.map hmmsearch_filt.map 119.165 -1 118.165 141 20:9 21:10 22:11 23:12 24:13 25:14 26:15
27:16 28:17 29:18 30:19 31:20 32:21 33:22 34:23 35:24 36:25 37:26 38:27 39:28 40:29 41:30 42:31 43:32 44:33 45:34 46:35 47:36 48:37 50:38 51:39 52:40 53:41 54:42 55:43 56:44 57:45 58:46 59:47 60:48 61:49 62:50 63:51 64:52 65:53 66:54 67:55 68:56 69:57 70:58 71:59 72:60 73:61 74:62 75:63 76:64 77:65 78:66 79:67 80:68 81:69 82:70 83:71
84:72 85:73 86:74 87:75 88:76 89:77 90:78 91:79 92:80 93:81 94:82 95:83 96:84 97:85 98:86 99:87 100:88 101:89 102:90 103:91 104:92 105:93 106:94 107:95 108:96 109:97 110:98 111:99 112:100 113:101 114:102 115:103 116:104 117:105 118:106 119:107 120:108 121:109 122:110 123:111 124:112 125:113 126:114 127:115 128:116 129:117 130:118 131:119 132:120 133:121 134:122 135:123 136:124 138:125 139:126 140:127 141:128 142:129 143:130 144:131 145:132 146:133 147:134 148:135 149:136 150:137 151:138 152:139 153:140 154:141 155:142 156:143 157:144 158:145 159:146 160:147 161:148 162:149
こんな感じで出力されます。最後のこの20:9 21:10というのが、要はこの2つのcontact mapの対応表となるわけです。これに準じてhmmsearch_filt.mapの番号を振り直すと……query側のCON 32 131 1.000がhmmsearch側のCON 21 119 1.000といった具合に、ほぼすべてのquery側の情報がhmmsearch側に含まれていることがわかりました!!
こうして、Uniref100データベースを用いたContact mapをクエリ配列に対応させることができるようになりました。
weight scoreの値について
GREMLINウェブサーバーのときに、Import to GREMLIN BETAのところで登場した
33_S 132_F 4.630 1.00
という行があるのですが、ここのScoreの4.630に相当する値は最後のmk_map.plを使ったときに表示されるProbの計算に使うので(GREMLINのWebページの解説にも出てる)、そのアウトプットファイルquery_filt.mapやhmmsearch_filt.mapを表示させる処理中で存在しているはずです。ということでmk_map.plを調べてみたら、score変数が隠されていました。それを再び表示するためには以下の修正をする必要があります。
mk_map.plを開き、48行目あたりにある次の箇所を修正します。
while(exists $CST[$c]){
my ($i,$j,$sc) = @{$CST[$c]};
my $prob = sprintf("%.3f",$N_v_H/(1+exp(-$N_v_S * ($sc - $N_v_B))) + $N_v_Y);
print MAP "CON $i $j $prob\n";
$c++;
}
これをこう
while(exists $CST[$c]){
my ($i,$j,$sc) = @{$CST[$c]};
my $prob = sprintf("%.3f",$N_v_H/(1+exp(-$N_v_S * ($sc - $N_v_B))) + $N_v_Y);
#print MAP "CON $i $j $prob\n";
# add $newsc to print score for Rosetta distance restraints
my $newsc = sprintf("%.3f", $sc);
print MAP "CON $i $j $newsc $prob\n";
$c++;
}
して、改めてmk_map.plを使ったcontactmapの計算を実行すると、例えば上のクエリ例のquery_filt.mapでは次のように表示されるようになります。
LEN 165
CON 32 131 4.774 1.000
CON 25 151 3.894 1.000
CON 133 141 3.490 1.000
CON 26 120 3.145 1.000
CON 59 127 3.017 1.000
CON 53 132 2.924 1.000
CON 25 113 2.892 1.000
まあGREMLINの結果とちょっとだけ値が少し違うんですけれど、これはデータベースで新しいものを使っているせいだと思いますたぶん。
これでscoreの値を得ることができました。必ずこの修正をしていたほうが良いです。
一括処理のシェルスクリプト
以下のファイルを一括でやりたい場合は、これらを単に連結するだけでいけるはずです。この処理をまとめたシェルスクリプトをcontactmap.shという名前で作成します。そしてコンタクトマップを予測したい配列の入ったFASTA形式のファイルをquery.faとして、 ./contactmap.sh query.fa とすれば実行されます。
順調に行けば1ファイルあたり40分〜80分程度で終了します。ファイル名が決めうちされている部分があるので、複数のfastaファイルを同一ディレクトリで実行するのはご遠慮ください。
後の一手のために、Process 9とProcess 10でPythonスクリプトによるファイル変換を行っています。これは https://github.com/BILAB/scripts にてスクリプトを公開していますので、ダウンロードしてお使いください。pymol_mapalign.py は次の項のPyMOL表示用プラグインを動作させる上で必要です。
# !/bin/sh
PROGNAME=$(basename $0)
query=$1
# set the path to uniclustDB and uniref100DB
uniclustDB="/Users/Agsmith/database/hhsuite/uniclust30_2018_08/uniclust30_2018_08"
uniref100DB="/Users/Agsmith/database/blast/db/FASTA/uniref100/uniref100"
# CPU number. Default is 12.
test ${PBS_NP} || PBS_NP=12
# main process
echo "Process 1 : hhblits to search for sequences"
hhblits -cpu ${PBS_NP} -i $query \
-d ${uniclustDB} \
-oa3m query.a3m \
-n 8 \
-e 1E-20 -neffmax 20 -nodiff -maxfilt 999999 -realign_max 999999 || exit 1
echo "Process 2 : filtering the sequences obtained from Proc 1"
hhfilter -i query.a3m -o query_filt.a3m -id 90 -cov 75 || exit 1
echo "Process 3 : reformat to a2m format to build hmm model using hmmbuild"
${HHLIB}/scripts/reformat.pl query_filt.a3m query_filt.a2m
hmmbuild --cpu ${PBS_NP} hmmout query_filt.a2m || exit 1
echo "Process 4: search for all similar sequences from uniref100 database and write in hmmsearch.a2m."
hmmsearch --cpu ${PBS_NP} -A hmmsearch.sto \
--incT 27 hmmout ${uniref100DB} > hmmsearch.out
echo "Process 5: filtering hmmsearch.sto"
${HHLIB}/scripts/reformat.pl hmmsearch.sto hmmsearch.a2m
hhfilter -i hmmsearch.a2m -o hmmsearch_filt.a3m -id 90 -cov 75 || exit 1
echo "Process 6-1: reformat hmmsearch_filt for CCMPred"
input=hmmsearch_filt
a3m2alncut.pl ${input}.a3m ${input}.aln ${input}.cut || exit 1
echo "Process 6-2: execute CCMPred to generate contact map for hmmsearch_filt"
ccmpred -t ${PBS_NP} -n 100 ${input}.aln ${input}.mtx || exit 1
input=hmmsearch_filt
mk_map.pl -aln ${input}.aln -chk ${input}.chk -cut ${input}.cut \
-mtx ${input}.mtx -map ${input}.map -do_apc
echo "Process 7-1: reformat query_filt for CCMPred"
input=query_filt
a3m2alncut.pl ${input}.a3m ${input}.aln ${input}.cut || exit 1
echo "Process 7-2: execute CCMPred to generate contact map for query_filt"
ccmpred -t ${PBS_NP} -n 100 ${input}.aln ${input}.mtx
input=query_filt
mk_map.pl -aln ${input}.aln -chk ${input}.chk -cut ${input}.cut \
-mtx ${input}.mtx -map ${input}.map -do_apc || exit 1
echo "Process 8: do map_align to merge the alignments"
map_align -a query_filt.map -b hmmsearch_filt.map > mapalignout.txt
echo "Process 9: renumbering the hmmsearch_filt.map using mapalignout.txt"
# pymol_mapalign.py is called by 'module load map_align'
pymol_mapalign.py hmmsearch_filt.map mapalignout.txt 1> hmmrenumbered.map 2>/dev/null
echo "Process 10: create distance restraint files for Rosetta AbinitioRelax"
# makecst.py is called by 'module load map_align'
makecst.py -f $query
PyMOL表示用プラグイン
この結果をPyMOLで表示するためのプラグインを作成しました。 https://github.com/BILAB/psicoに便利なPyMOLプラグインを入れております。この中のcontactmap.pyが表示プラグインです。
PyMOLプラグインが入っているpsicoディレクトリの使い方は、まず
git clone https://github.com/BILAB/psico.git
コマンドで中のpsicoディレクトリを適当な場所にダウンロードしたあと、これについてのPATHをPyMOL側から通しておくことです。例えば~/appsにpsicoディレクトリを生成した場合は、~/.pymolrcを作成して
# ~/appsはpsicoディレクトリの場所に合わせる
sys.path.append(os.path.expanduser('~/apps'))
import psico.fullinit
としておきます。この状態でPyMOLを立ち上げれば、contactmapを始めとしたpsicoの中のプラグインに由来する追加コマンドが使えるようになっているはずです。
PyMOLの窓にhelp contactmapとすると、使い方が表示されます。
PyMOL>help contactmap
DESCRIPTION
visualize the results from map_align algorithm in PyMOL.
https://github.com/sokrypton/map_align
ARGUMENTS
selection = string: atom selection {default: all}
chain = string: chain ID you want to map {default: ''}
align = string: name of map_align result txt file {default: 'hmmrenumbered.map'}
pcut = float: probability cutoff to display {default: 0.999}
offset = int: slide the sequence number to fit its original number manually {default: 0}
EXAMPLE
contactmap 5dir, A, /path/to/hmmrenumbered.map, offset=-7
contactmap <object name>, <chain ID>とし、上記Process 9で作成したhmmrenumbered.mapのパスを適切に入れてやると動きます。pcutは表示させるときのProbabilityのカットオフ値です(Probabilityの低いものまで全部表示させていると大変なので)。offsetは、PDBの番号とFASTAのナンバリングが合わない時にのみ、適当な整数値を与えてやってください。以下の図はPDB: 5DIRでやったときの図です。PDBのナンバリングととquery.faの残基数は7ずれていたため、offset=-7にしてあります。
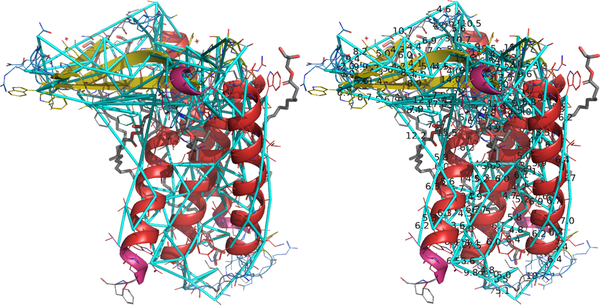
こうしてみると、だいたいのCβ-Cβ距離は6~8 Å以内にあることがわかります。
特性
GREMLIN/CCMPredは以下のような特性があります。
- 配列空間が大きければ大きいほど、精度が上昇する:つまり、タンパク質をコードするアミノ酸配列データベースが増えれば増えるほど、精度が上がっていきます。現在も配列空間はどんどん更新されており、Uniref100に集約されていますので、新しいUniref100データベースを利用すれば精度が増す仕様となっています。
-
クエリ配列に対する類縁配列が多いほど精度が上昇する:
hhblitsなどでマルチプルシーケンスアラインメントが取れた本数だけ精度が上がる仕組みになっています。類縁配列が乏しいタンパク質だとよい精度が出ません。