MSCIコンペについて既に振り返りはしたが、今回のコンペでは「どのようにして最終サブミットのためのモデルを作成するか」が非常に重要な要素でずっと頭を悩ませていたので、これについて考えていこうと思う。加えて自分の特徴量エンジニアリングの手法についても備忘として残しておく。
CV と Public LB と Private LB
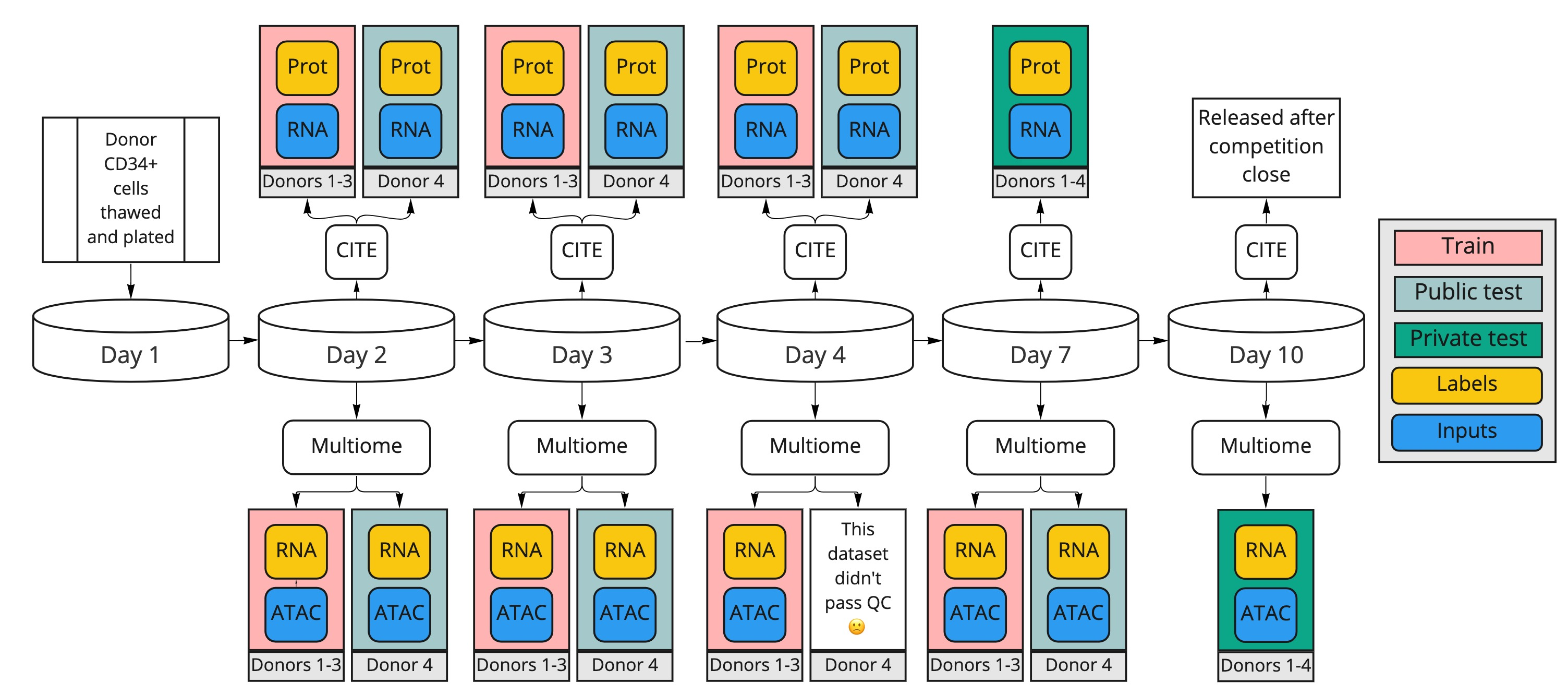
今回のコンペでは、CVでのデータの分布とLBでのデータの分布が異なっていた。CITE に限定して考えていく。
- TrainデータはDay2,3,4 (Donor1-3)の細胞群 (図では赤色)
- Public LBデータはDay2,3,4 (Donor4)の細胞群 (図では水色)
- Private LBデータはDay7 (Donor1-4)の細胞群 (図では緑色)
このように、Public LBにはTrainにはない未知のDonorの情報が、Private LBには未知のDay・未知のDonorの情報が含まれていた。そのため、Public LBでは未知のDonorをうまく予測できるモデルを、Private LBでは未知のDayをうまく予測できるモデル (Privateでは未知のDonorより未知のDayの方がデータ数が多いため) が必要であると考えていた。
7th place の手法
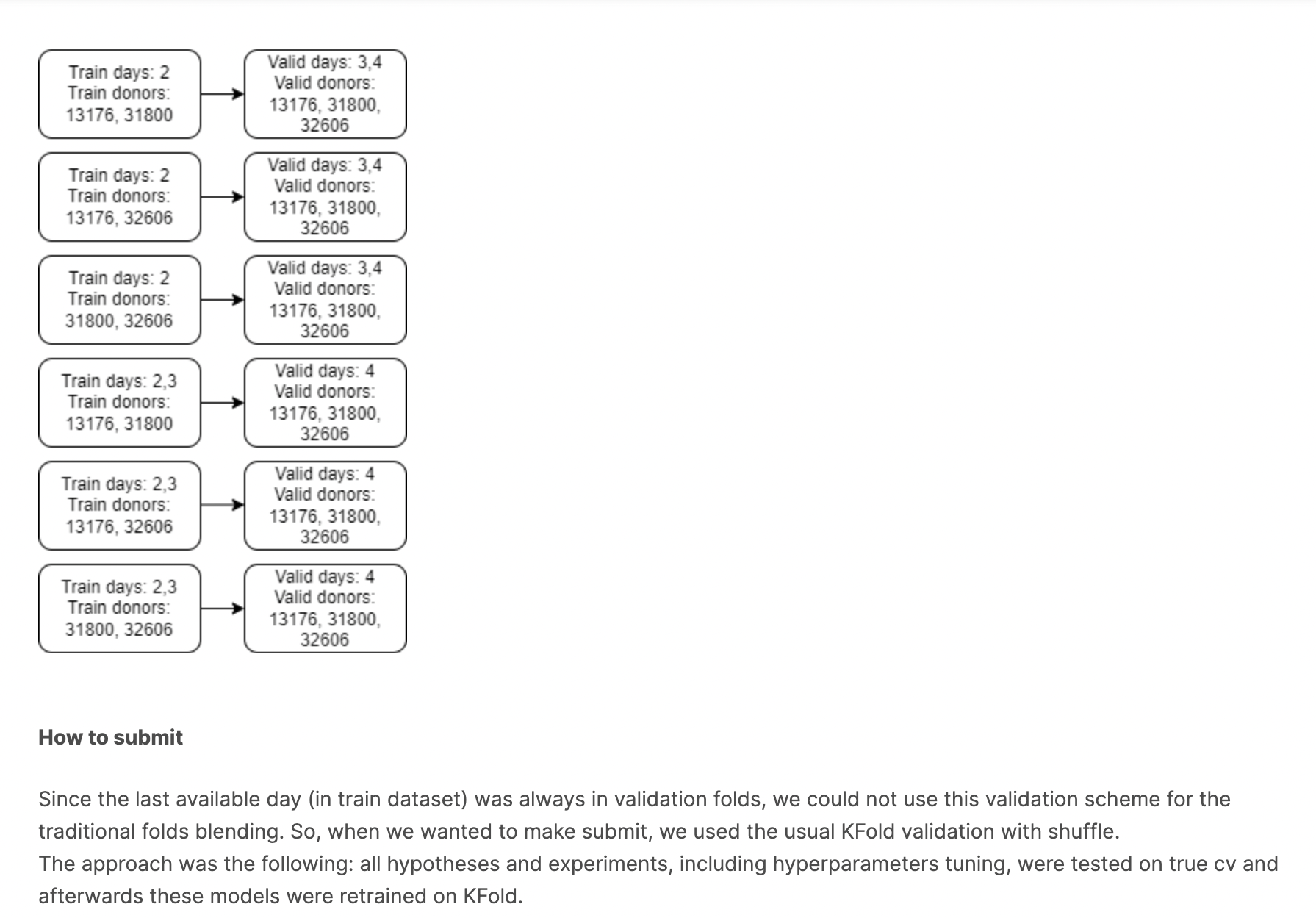
この人たちは、CVの時には以下のようにPrivate LBを完全に再現して実験を進めていた。未知のDayとしては未来のDayをvalidに使ったり、Donorに関しても3人のvalidのうち1人だけが未知のDonorであるという徹底ぶり。ただ、これだとSubmitをする時に問題が起こる。CVで作成した6つのモデルを用いてSubmitを作成しようとすると、どうしてもDay3やDay4のデータを使うことができない。Day7を予測するためにはDay3やDay4のデータが有効であろうことは確実なので、これらを使わない選択肢はない。
そこで彼らはSubmitにはKFoldを使用した。全ての仮説検証や特徴量作成、パラメータチューニングは上のCVでおこない、最後のSubmitのみでKFoldを使用したらしい。おそらく、以下のPrivateを模倣したFoldにおいて、By donor,By day,Kfoldを試した結果、ベストのスコアが出たものがKfoldだったのだろう。どのCVモデルがうまくPrivateを予測できるかの答えなどなくて、結局Privateを模倣して実際に実験することで初めて分かることなのだろう。何となく自分は未知のDonorを予測するならBy donorのCVモデルを使って、未知のDayを予測するならBy dayのCVモデルを使うと思い込んでいたけど、明確な理由はなかった。実際に間違っていたし、明確な理由など見つかるはずもなかった。自分は割と理論ベースで話を進めようとしているけど、実際に試すことの方がより重要で説得力がある。
3rd place の手法
この人の特徴的な点は、adversarial validation を使用していたところ。これは学習データとテストデータを結合し、テストデータか否かを目的変数とする二値分類を行うことで、学習データとテストデータの分布が同じかどうかを判断する手法である。結果として、今回のコンペのPrivate LBデータとその他のデータを高精度で分類することができていた (つまり、Private LBとその他の分布が明確に異なっていたということ)。そのため、本コンペではPublic LBを信用すべきではなかったといえる。
ここからが重要な部分。この人は学習データの中のPrivate LBに近い10%をvalidationデータとして使用したのだ。そして、CVでの最高の値がPrivate CVでの最高の値であったようだ。確かに言われてみれば割と単純な話だが、実際に確かめて実装するとなるとそう簡単にはいかないと思う。ただ、この手法はどのコンペでも有効に働いてくれそうなので、実装できるようになっておきたい。
2nd place の手法
コンペ中常に1位を独走してきたSenkinさんの手法。彼は一旦は Random Kfold をおこなっていた。CV (Donor1,2,3)とPublic LB (Donor4)の相関がうまく取れていたため、Donorの違いによる影響はないと結論づけた。自分にはこの考察ができていなかった。CVの意味だったり分割方法をもっと考える必要があると痛感している。
この後に、Dayの違いによる影響をGroup Kfold by dayを用いて検討した。具体的にはそれぞれの特徴量がCVスコアを向上させるかを検討した。だが、最終サブミットにはこのCVは使わずに、Kfoldを使用したようだ。7th place と同じ手法と言える。
まとめ
今回のコンペでは、仮説検証の段階ではGroup Kfoldを使用して、Random Kfoldで最終サブミットのモデルを作成している人が大半であったと思う。興味深い点は、GroupKfoldで作成した特徴量やパラメータをそのまま使用していたことだ。何となくCVの枠組みによってパラメータとかは変わりそうな気がするけど、実際にはGroup Kfoldで高いスコアが出た特徴量やパラメータは、Random Kfoldでも高いスコアが出るようだ。これは今後のコンペでも役立つ知識だと思う。
(備忘) 特徴量エンジニアリング
データ概要
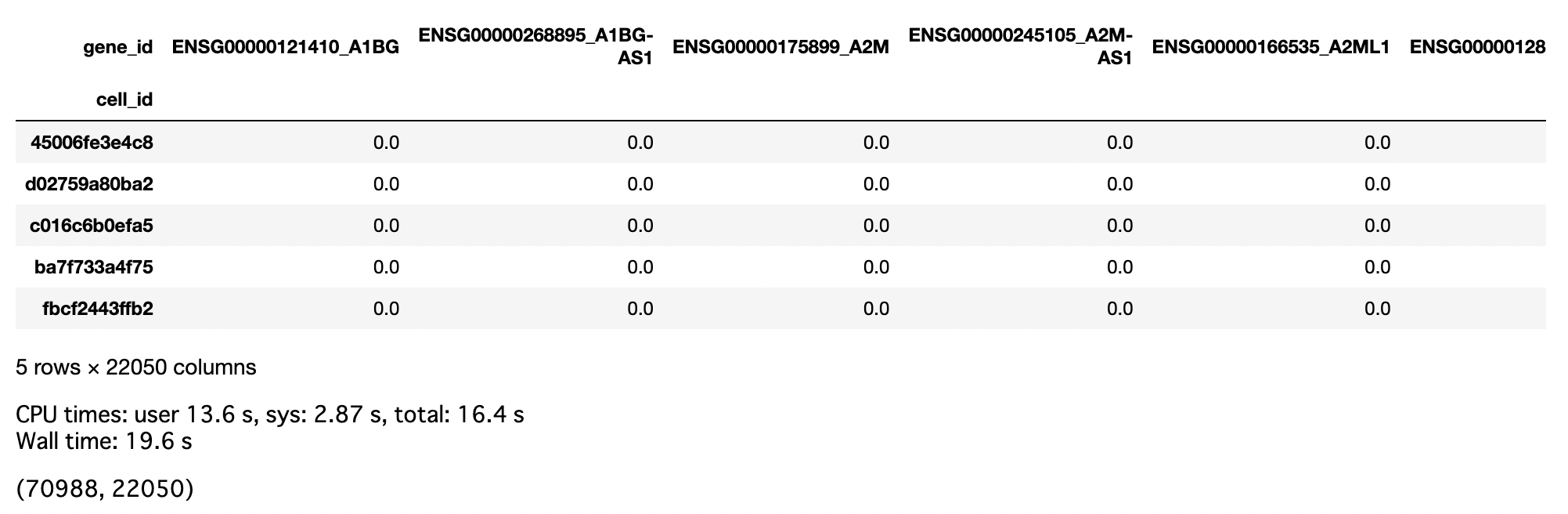
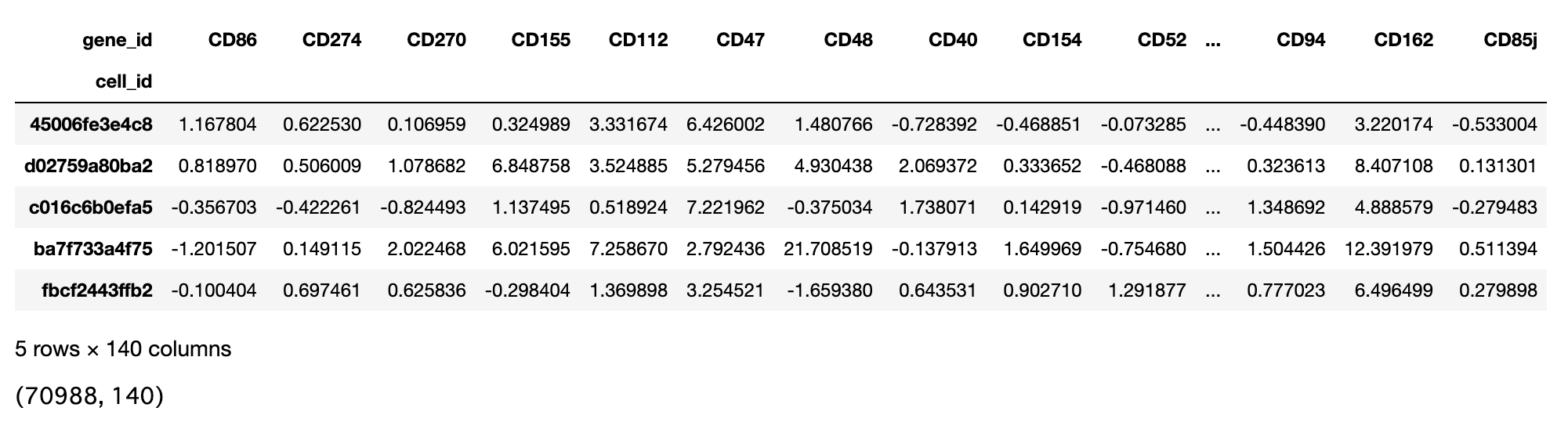
データ概要は以下に示す通りである。一つ目に示したものが訓練データで、行には各細胞が、列には遺伝子名が記されている。二つ目に示したものがテストデータで、行には訓練データと同様に細胞が、そして列には目的変数となるタンパク質名が記されている。22050個ある特徴量をいかに処理するかが本コンペの醍醐味だったと思う。
自分の解法
-
train, testの全ての細胞において値が一定である特徴量を削除する。
ある特徴量が両者において値が一定である(今回は全て0)場合には、当然であるがその特徴量は予測には役立たない。仮にタイタニックの乗客が全員男だったとしたら、性別という特徴量は無駄なわけだから。このような無駄な特徴量が予測に悪影響を及ぼす可能性もあるので、これらは削除するべき。 -
tSVDによる次元圧縮
前述のように元々の特徴量が22050あり処理が難しいので、特異値分解による次元圧縮をおこなった。特異値分解をすることによって、データを構成するのに大きな影響を与えるベクトルから順に列ベクトルに割り当てられる。影響力の小さいベクトルを消去することによって情報を保持したまま次元圧縮をすることができる。

参考
https://qiita.com/sakami/items/d01fa353b4e1f48623a8
https://qiita.com/K_Noguchi/items/60ba6a08d1f913e74a83
https://thinkit.co.jp/article/16884 -
重要な遺伝子を特定する
目的変数であるタンパク質(testにおけるCD86など)の予測に重要と考えられる遺伝子(trainにおけるENSG...)を特定した。遺伝子はタンパク質の「設計図」のようなものであり、どの遺伝子がどのタンパク質の生成に寄与しているかはある程度判明している。その組(1対1でない場合もある)をwikipeidaやEnsemble genome browserを利用して一つ一つ自分で調べていった。これらの「重要な」遺伝子には次元圧縮によって情報を失ってほしくないので、次元圧縮後のデータに特徴量としてそのまま追加した。 -
細胞種を特徴量に追加
各細胞がどの分化ステージにいるかを明示してあげることで、タンパク質発現の予測が容易になると考えた。One hot encodingにより特徴量を追加した。