※split関数の書式を修正しました(2023.12.02)
##3原子の分子動力学計算
3つの原子だけを扱う分子動力学(MD)コードを書いてみる。
ステップバイステップでMDっぽくしていく。
###0. 準備
Tkinterをインストールしておく必要がある。
WindowsではPythonパッケージのインストールの際に tcl/tk and IDLE にチェックを入れておくことで導入される。
Linuxの場合、例えばubuntuでは
$ sudo apt install python3-tk
とすればよい。
###1. キャンバスに原子を配置する
原子3つの初期配置と速度をファイルに書いておく。
15 10 0 0
20 10 0 0
20 15 0 0
initial. d の各行は 原子のx座標、y座標、速度のx成分、y成分 の4つの項目からなり、スペースで区切られている必要がある。
物体を描画できるようにキャンバスを開き、ファイル(initial.d)から原子配置(と速度)情報を読み取って原子を初期位置に配置するプログラム例を下に示す。
後々のため、ダミーのボタンも用意している(ここではボタンが押されても特に何もしない)。
ここではinitial.dが3行なので原子数は3つとなるが、行数を増やすことで原子数が増やせる。プログラム中では各行を読み込む度に配列サイズを増やしている。
座標の単位はÅ、速度の単位はÅ/sとしている。
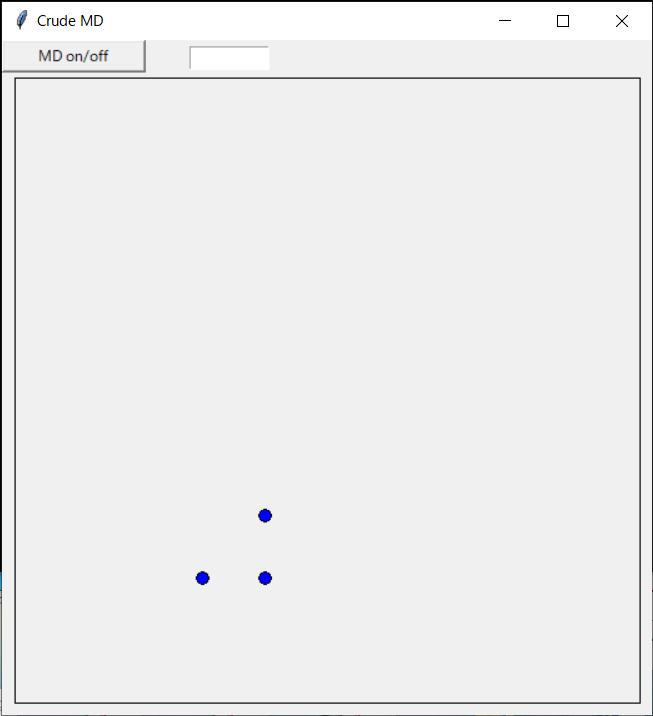
プログラム例を示す。
import tkinter as tk
def dummy(event):
print('button is pressed')
def drawatom(x,y):
scl=10
rad=5
x1=x0+x*scl
y1=y0+l-y*scl
canvas.create_oval(x1-rad,y1-rad,x1+rad,y1+rad, fill='blue')
# creation of main window and canvas
win = tk.Tk()
win.title("Crude MD")
win.geometry("520x540")
x0=10
y0=10
l=500
canvas = tk.Canvas(win,width=l+x0*2,height=l+y0*2)
canvas.place(x=0, y=20)
canvas.create_rectangle(x0,y0,l+x0,l+y0)
# declaration of arrays
rx = []
ry = []
vx = []
vy = []
fx = []
fy = []
# button sample (dummy)
button_dummy = tk.Button(win,text="dummy",width=15)
button_dummy.bind("<Button-1>",dummy)
button_dummy.place(x=0,y=0)
# read initial position and velocity
f = open('initial.d', 'r')
i = 0
for line in f:
xy = line.split()
rx = rx + [float(xy[0])]
ry = ry + [float(xy[1])]
vx = vx + [float(xy[2])]
vy = vy + [float(xy[3])]
print(rx)
drawatom(rx[i],ry[i])
i = i + 1
win.mainloop()
###2. ボタンイベントを取得して原子を移動
上記のプログラムを元に機能を追加。
ボタンが押されたイベントを検出して、原子を斜め上方向にただ連続的に動かすようにしたもの。
その際,原子を動かすごとにステップ数(step)をカウントアップしていき、ステップ数をウインドウ上部に表示する機能も付与している。
import tkinter as tk
def dummy(event):
global imd # to substitute value into variable
if (imd == 0 ):
imd = 1
else:
imd = 0
def drawatom(x,y):
x1=x0+x*scl
y1=y0+l-y*scl
return canvas.create_oval(x1-rad,y1-rad,x1+rad,y1+rad, fill='blue')
def moveatom(obj,x,y):
x1=x0+x*scl
y1=y0+l-y*scl
canvas.coords(obj,x1-rad,y1-rad,x1+rad,y1+rad)
# creation of main window and canvas
win = tk.Tk()
win.title("Crude MD")
win.geometry("520x540")
x0=10 # Origin
y0=10 # Origin
l=500 # Size of canvas
scl=10 # Scaling (magnification) factor
rad=5 # Radius of sphere
canvas = tk.Canvas(win,width=l+x0*2,height=l+y0*2)
canvas.place(x=0, y=20)
canvas.create_rectangle(x0,y0,l+x0,l+y0)
imd = 0 # MD on/off
# declaration of arrays
rx = []
ry = []
vx = []
vy = []
fx = []
fy = []
obj = []
# button sample (dummy)
button_dummy = tk.Button(win,text="dummy",width=15)
button_dummy.bind("<Button-1>",dummy)
button_dummy.place(x=0,y=0)
# Entry box (MD step)
entry_step = tk.Entry(width=10)
entry_step.place(x=150,y=5)
# read initial position and velocity
f = open('initial.d', 'r')
n = 0
for line in f:
xy = line.split()
rx = rx + [float(xy[0])]
ry = ry + [float(xy[1])]
vx = vx + [float(xy[2])]
vy = vy + [float(xy[3])]
print(rx)
obj = obj + [drawatom(rx[n],ry[n])] # obj[] holds pointer to object
n = n + 1 # number of total atoms
print("number of atoms = ",n)
step = 0
stepend = 1000
while step < stepend:
if imd == 1:
for i in range(n):
rx[i] = rx[i] + 0.01
ry[i] = ry[i] + 0.01
moveatom(obj[i],rx[i],ry[i])
step = step + 1
entry_step.delete(0,tk.END)
entry_step.insert(tk.END,step)
win.update()
win.mainloop()
###3. ボタンが押されたらMDをオン・オフする
上記をさらに改変。
ボタンが押されたらMDのon/offを切り替えるようにした。
原子同士の相互作用をMorseポテンシャルで計算し、Verlet法で原子を動かすようになっている。
import tkinter as tk
import math
def dummy(event):
global imd # to substitute value into variable
if (imd == 0 ):
imd = 1
else:
imd = 0
def drawatom(x,y):
x1=x0+x*scl
y1=y0+l-y*scl
return canvas.create_oval(x1-rad,y1-rad,x1+rad,y1+rad, fill='blue')
def moveatom(obj,x,y):
x1=x0+x*scl
y1=y0+l-y*scl
canvas.coords(obj,x1-rad,y1-rad,x1+rad,y1+rad)
def v(rang):
ep = 0.2703
al = 1.1646
ro = 3.253
ev = 1.6021892e-19
return ep*(math.exp(-2.0*al*(rang-ro))-2.0*math.exp(-al*(rang-ro))) *ev
def vp(rang):
ep = 0.2703
al = 1.1646
ro = 3.253
ev = 1.6021892e-19
return -2.0*al*ep*(math.exp(-2.0*al*(rang-ro))-math.exp(-al*(rang-ro))) *ev*1.0e10
# creation of main window and canvas
win = tk.Tk()
win.title("Crude MD")
win.geometry("520x540")
x0=10 # Origin
y0=10 # Origin
l=500 # Size of canvas
scl=10 # Scaling (magnification) factor
rad=5 # Radius of sphere
canvas = tk.Canvas(win,width=l+x0*2,height=l+y0*2)
canvas.place(x=0, y=20)
canvas.create_rectangle(x0,y0,l+x0,l+y0)
imd = 0 # MD on/off
# declaration of arrays
rx = [] # ang
ry = []
vx = [] # ang/s
vy = []
fx = [] # N
fy = []
epot = []
obj = []
dt = 1.0e-16 # s
wm = 1.67e-37 # 1e-10 kg
# button sample (dummy)
button_dummy = tk.Button(win,text="MD on/off",width=15)
button_dummy.bind("<Button-1>",dummy)
button_dummy.place(x=0,y=0)
# Entry box (MD step)
entry_step = tk.Entry(width=10)
entry_step.place(x=150,y=5)
# read initial position and velocity
f = open('initial.d', 'r')
n = 0
for line in f:
xy = line.split()
rx = rx + [float(xy[0])]
ry = ry + [float(xy[1])]
vx = vx + [float(xy[2])]
vy = vy + [float(xy[3])]
fx = fx + [0]
fy = fy + [0]
epot = epot + [0]
obj = obj + [drawatom(rx[n],ry[n])] # obj[] holds pointer to object
n = n + 1 # number of total atoms
print("number of atoms = ",n)
step = 0
stepend = 100000
while step < stepend:
if imd == 1:
# Verlet(1)
for i in range(n):
rx[i] = rx[i] + dt * vx[i] + (dt*dt/2.0) * fx[i] / wm
ry[i] = ry[i] + dt * vy[i] + (dt*dt/2.0) * fy[i] / wm
vx[i] = vx[i] + dt/2.0 * fx[i] / wm
vy[i] = vy[i] + dt/2.0 * fy[i] / wm
# Force and energy
for i in range(n):
fx[i] = 0
fy[i] = 0
epot[i] = 0
for i in range(n):
for j in range(n):
if (i != j):
rr = math.sqrt((rx[i]-rx[j])**2 + (ry[i]-ry[j])**2)
drx = rx[i] - rx[j]
dry = ry[i] - ry[j]
fx[i] = fx[i]-vp(rr)/rr*drx
fy[i] = fy[i]-vp(rr)/rr*dry
epot[i] = epot[i]+v(rr)/2.0
# Verlet(2)
for i in range(n):
vx[i] = vx[i] + dt/2.0 * fx[i] / wm
vy[i] = vy[i] + dt/2.0 * fy[i] / wm
moveatom(obj[i],rx[i],ry[i])
step = step + 1
entry_step.delete(0,tk.END)
entry_step.insert(tk.END,step)
win.update()
win.mainloop()