Genome Sequencing in a Nutshell - The Databricks Blogの翻訳です。
本書は抄訳であり内容の正確性を保証するものではありません。正確な内容に関しては原文を参照ください。
2016年の記事です。
これはNorthwest Genome CenterとUniversity of WashingtonのDeborah SiegelとDatabricksのDenny LeeによるSparkを用いたADAMによるゲノム変異体解析のコラボレーションから生まれたゲスト記事です。
これはK-Means、ADAM、Apache Sparkを用いたゲノム変異体解析に関する3パートのシリーズのパート1です。
イントロダクション
過去数年間を通じて、我々はゲノムシーケンシングのコストと時間の急激な削減を目撃してきました。ゲノムシーケンスのバリエーションを理解することのポテンシャルは、一般的な病気の傾向がある人の特定、希少疾病の解決、医師による個人にパーソナライズした処方と投薬の実現と多岐にわたります。
この3パートのブログ記事では、ゲノムシーケンシングの入門書とそのポテンシャルをご説明します。ここでは、ゲノムシーケンスの差異を見るゲノム変異体解析にフォーカスし、Databricksコミュニティエディションを用い、Apache SparkとADAMを活用することでどのように高速に行えるのかを説明します。最後に、ゲノム変異データに対してk-meansアルゴリズムを実行し、これらの変異体に基づいて個人の起源となる地理グループを予測するモデルを構築します。
最初の記事では、ゲノムシーケンスの入門を説明します。また、並列バイオインフォマティック解析にフォーカスしている2つ目の記事ゲノム変異体解析の並列化、3つ目の記事ゲノム変異体とK-Meansを用いた地域グループの予測を読み進めることもできます。
ゲノムシーケンシング
非常にシンプルな言語のアナロジー
30億の文字、他の文字と入り混じって存在する約25,000の単語を含む長い文字列を想像してみます。単語のいくつかは文も構成しています。文字列の変更、追加、削除、文字列のグルーピングは、単語や文の構造や意味を変更することがあり得ます。

それぞれの長い文字列には、このような差異を引き起こす場所が約1000万から3000万存在しています。そして、これが面白くなります。もちろん、すべてはもっと複雑なものです。しかし、これはゲノムデータに対する有用な抽象表現を説明しています。
ゲノムにおいては、我々は文字列(塩基)のどこに単語(遺伝子)が配置されるのかに関する知識を構築してきており、違いがある場所(変異体)がどこなのかを発見してきました。しかし、我々が全てを理解しているわけではありません。いまだに我々は変異体の効果が何であるのか、遺伝子は互いにどのような関係があるのか、これらが特定の状況下で別の形態、別の量で発現しうるのかを学んでいる最中です。

生物学か外国語かわからん
つまりゲノムシーケンシングとは
ゲノムシーケンシングには、化学とゲノムをコーディングする文字列(A,G,C,T)を順番で読み取るための記録技術が関与します。
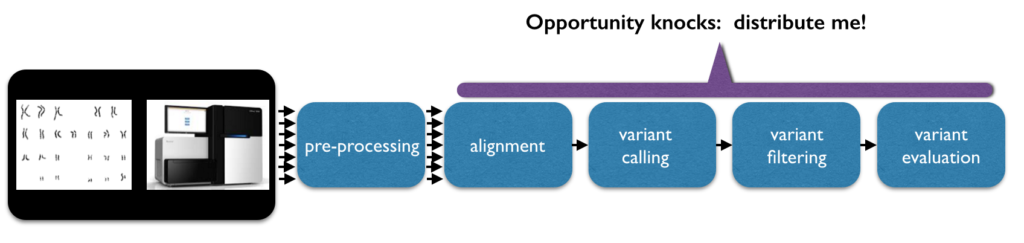
最初にデータは短い文字列の形で読み込まれます。ある個人のゲノムの30x(ゲノム全体が平均30回シーケンスされる)のカバレッジ(30xは一般的なゴールです)に対して、150文字ごとの約6億の短い文字列が存在し得ます。データの前処理においては、文字列は通常はリファレンスシーケンスにマッピング/調整されます。最終的には、これによって全ての塩基に対して定義された位置を提供します。割り当てられたシーケンスされたデータの変異体解析によって、シーケンスとリファレンスシーケンスや他に割り当てられたシーケンスを比較することでコードの違いを見つけ出し、ある個人の変異体に遺伝子型を割り当てます。

検知された変異体のいくつかはノイズによるものであり、カバレッジ、品質、ドメイン固有のバイアスのようなパラメーターに対する厳密な閾値によってフィルタリングすることができます。ハード(コードされた)なフィルタリングを行うのではなく、いくつかの解析では、ガウジアン混合モデルをフィッテングすることで変異体をフィルタリングします。さらに後段の解析では、アナリストはデータを計測、探索し、(与えられた入力サイズでは数が少ない)より重要な変異体を識別しようとし、どのような機能的な効果があるのかを予測しようとします。
なぜシーケンスなのか?
データサイエンスの観点において、ゲノムシーケンス(そして、サブセットであるエクソームも)は興味深いデータと言えます。長い時間を経てコードがどのように、そしてなぜ進化したのかに関するヒントを得るためにシーケンスに対する知識を活用することができます。ゲノムシーケンシングの研究から得られる知識は、これまで以上に医療に組み込まれています。今では、ゲノムシーケンシングはnon-invasive prenatal diagnostics:無侵襲的出生前検査に使われています。まもなく、ゲノムシーケンシングが医療スクリーニングや診断試験に使われるようになり、ゲノム医療を拡張するための取り組みが行われています。
研究、発見側においては、自閉症、心臓疾患、特定の腫瘍のような一般疾病の傾向がありそうな人の変異体のパターンや変異体を見つけるための、大規模グループ、人口規模のゲノムシーケンシングの研究が行われています。また、シーケンシングの研究は、薬品の代謝に影響を及ぼす変異体も導き出しており、医師が個人ごとにパーソナライスした処方、投薬を行えるようになっています。希少な遺伝性疾病の場合、特定の家族のシーケンスによって原因となる変異体の発見に繋がるケースが多くなっています。

(画像の提供元: Frederic Reinierから許可を得て)
過去五年間において、シーケンシングの実験によって、数百の希少疾病と遺伝変異体を紐付けました。
「個々を見た場合、ある希少疾病は一握りの家族にのみ影響を与えるだけかもしれません。全体的に見るとアメリカだけでも、希少疾病は2000万から3000万の人々にインパクトを与えます。」
これらの理由から、シーケンスの読み取りと解析を推し進めるリソースが確保されています。イギリスのNational Health Serviceは2017年までに希少疾病や癌を持つ人がいる家族の10万のゲノムをシーケンスするためのプロジェクトを進めています。アメリカでは、National Human Genome Research Institute (NHGRI)が次の4年間で4000万ドルを希少疾病の研究に、2億4千万ドルを一般疾病の研究にファンディングすることを計画しています。また、バイオインフォマティクスをスケールし、RNA-seq、マイクロバイオームのシーケンス、免疫システムと癌プロファイルのシーケンスのように、大量のシーケンスデータに対してデータサイエンスを適用する障壁を引き下げる努力によってメリットが得られるシーケンスも存在します。
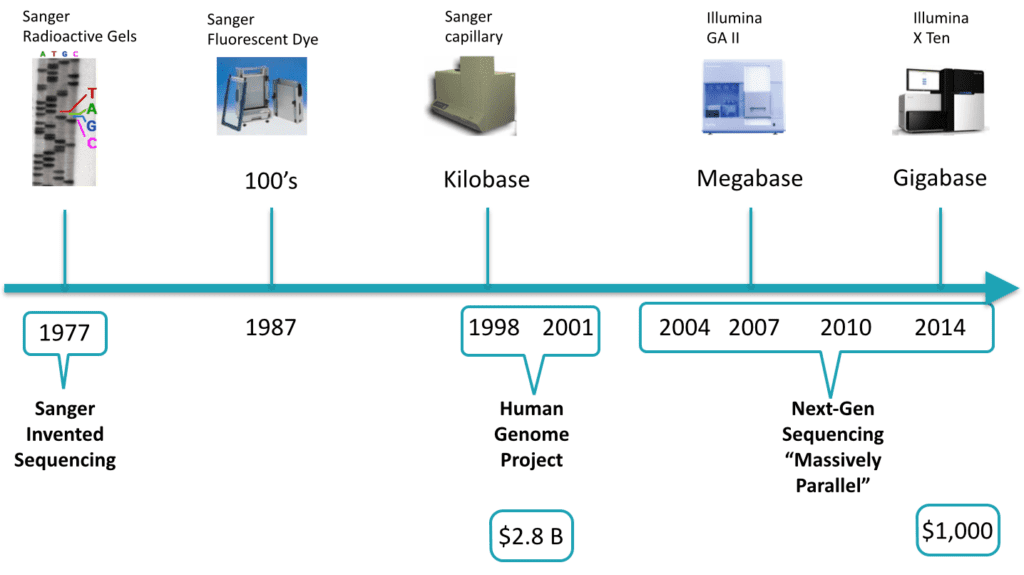
シーケンシングの技術は、加速して成長し続けています。1998年と2001の間に、最初の人間のゲノムがシーケンスされました。2009年時点ではこれは28億ドルかかりました。今では、1000ドル程度で3日でゲノムがシーケンスされます(詳細は、National Institutes of Health: National Human Genome Research Institute > DNA Sequencing Costsをご覧ください)。シーケンスの実験の最初の25年においては、化学者のみが一筋のDNAのみをシーケンスすることしかできず、非常に労力を要し、遅く、高価なものとなっていました。次世代シーケンシングは高度に並列化され、同じ実験で複数のDNAをシーケンスすることが可能となりました。また、分子インデックスを用いることで、複数の人のDNAを一緒にシーケンスすることができ、解析の際にデータを分離することが可能となりました。それほど遠くない未来に、オプトインした地球上の多くの人々のゲノムがシーケンスされるだろうということは、それほど非現実的な話ではありません。次世代シーケンシングの詳細に関しては、Coming of age: ten years of next-generation sequencing technologiesをご覧ください。
アプリケーションと環境によりますが、現在のシーケンシングの装置は一日あたり600ギガベースを読み込むことができます。中規模から大規模のシーケンシング組織では、同時に実行できるこのような装置を複数持っています。のちほど詳細を見ていきますが、バイオインフォマティクスが直面している課題の一つは、変異体を解析する後段のソフトウェアがこれまではデータモデル自身ではなく、固有かつ拡張性のないフォーマットに最適化されているということです。結果として、パイプラインの脆さが存在し、スケーラビリティの障害となっていました。今では、高度に並列なシーケンシングを行なっており、多くの人が並列バイオインフォマティクス解析の方向に進んでいます。
公開データ
ゲノムシーケンスデータは一般的にプライベートなものです。2007年と2013年の間は、1000のゲノムプロジェクトはパブリックの「人口規模のシーケンス」のための初期の取り組みの状況にありました。最終フェーズまでには、26グループの2504人に対するシーケンスのカバレッジのいくつかの部分を提供しています。Databricksコミュニティエディションでノートブックを開発するためのリソースとして、このプロジェクトから容易にアクセスできるデータを使用しました。
次のステップ
次の記事ゲノム変異体解析の並列化では、並列バイオインフォマティクス解析を見ていきます。これもスキップして、ゲノム変異体とK-Meansを用いた地域グループの予測を読むこともできます。
リソース
ノートブックを作成する助けになった以下のリソースを特筆しておきたいと思います。
- ADAM: Genomics Formats and Processing Patterns for Cloud Scale Computing (Berkeley AMPLab)
- Andy PetrellaのLightning Fast Genomics with Spark and ADAMと関連のGitHub repo
- Neil FergusonのPopulation Stratification Analysis on Genomics Data Using Deep Learning
- Matthew ConlenのLightning-Viz project
- (Sparkによるゲノミクスにおける)Timothy Danford’s SlideShare presentations
- Centers for Mendelian Genomics uncovering the genomic basis of hundreds of rare conditions
- NIH genome sequencing program targets the genomic bases of common, rare disease
- The 1000 Genomes Project
また、Anthony Joseph、Xiangrui Meng、Hossein Falaki、Tim Hunterによる追加の貢献とレビューに感謝します。