Simplifying Genomics Pipelines at Scale with Databricks Delta - The Databricks Blogの翻訳です。
サンプルノートブックはこちらからダウンロードできます。
2003年のヒューマンゲノムプロジェクトの完了以来、当初は30億ドル1かかっていたDNAシーケンスの費用が今では1000ドルとなっており、劇的なコスト削減によって大量のデータが生まれています。

ソース: DNA Sequencing Costs: Data
結果として、企業がDNAシーケンシングを人口規模で実行できるステージにまでゲノミクスの分野は成熟しました。しかし、DNAコードのシーケンシングは最初のステップであり、生データは分析に適したフォーマットに変換される必要があります。一般的にこの作業は、単一のノードで一度に一つのサンプルに対して一連のバイオインフォマティクスツールと、カスタムスクリプトと処理を適用し、遺伝的変異体コレクションを取得することで完了します。バイオインフォマティクスサイエンティストはこれらのパイプラインの構築に多くの時間を費やしています。ゲノミクスデータセットはペタバイト規模にまで拡大しており、以下のようなシンプルな質問に迅速に応えることも難しくなってきています。
- 今月どれだけのサンプルをシーケンシングしたのか?
- 固有の変異体をいくつ検知したのか?
- 異なる変異クラスにおいていくつの変異体を見たのか?
さらに問題を複雑にしているのは、アクセス、検索可能な状態で数千の個人からのデータを蓄積、追跡、バージョン管理ができないということです。結果として、研究者は分析を行う際にゲノムデータのサブセットを複製することになり、全体的なストレージ容量とコストを引き上げることになります。この問題を緩和するために、研究者は多くの場合、6ヶ月から2年間の「データフリーズ」の戦略をとっています。この期間は新しいデータに対する作業を停止し、既存の「氷漬けされた」データのコピーにフォーカスします。短い時間間隔で増分的に分析を積み上げるソリューションが存在しないため、研究プロセスが遅延しています。
サイエンティストがデータを探索でき、分析パイプラインに対して繰り返し作業を行い、新たな洞察を得られる柔軟性を保ちつつも、業界規模のゲノムデータを業界規模で処理できる頑健性のあるソフトウェアが求められています。
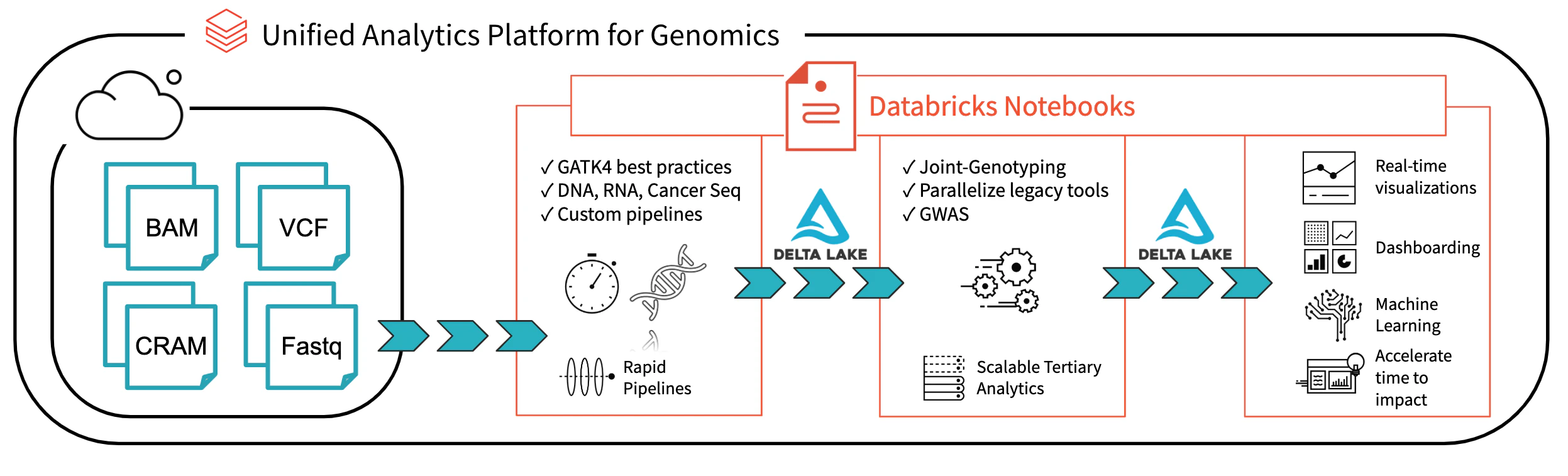
図1:Databricksにおけるエンドツーエンドのゲノム分析アーキテクチャ
Databricks Delta Lake:リアルタイムビッグデータ分析統合管理システムを用いることで、Databricksのレイクハウスプラットフォームは、現在研究者が直面しているデータガバナンス、データアクセス、データ分析の問題を解決するための大きな一歩を踏み出しました。Databricks Delta Lakeを用いることで、すべてのゲノムデータを一箇所に格納し、新たなデータが登録されるたびにリアルタイムで分析を実行することができます。Unified Analytics Platform for Genomics(UAP4G)のゲノムファイルフォーマットの読み取り、書き込み、処理に関する最適化技術と組み合わせることで、我々はゲノミクスパイプラインのワークフローに対するエンドツーエンドのソリューションを提供します。UAP4Gアーキテクチャは、お客様自身のパイプラインにプラグインでき、お客様の高度な分析を実行できる柔軟性を提供します。例としては、以下の品質管理メトリクスを表示するダッシュボードと、お客様固有の要件に応じてカスタマイズでき、自動で計算表示されるビジュアライゼーションをハイライトしています。
この記事の残りの部分では、上述の品質管理ダッシュボードを作成するステップをご説明します。このダッシュボードはサンプルの処理が終わるとリアルタイムで更新されます。ゲノムデータを処理するためにDeltaベースのパイプラインを使用することで、お客様はリアルタイム、サンプルごとの可視性を持つパイプラインを操作することができます。Databricksのノートブック(およびGitHub、MLflowと連携することで)を用いることで、結果の再現性を確保できるように分析を追跡、バージョン管理ができるようになります。バイオインフォマティクス専門家がパイプラインの維持に割く時間を削減でき、より多くの時間を発見に費やせるようになります。我々はUAP4Gを、アドホック分析からプロダクション規模でのゲノミクスへの変換を推進するエンジンと考えており、これによってさらに遺伝学と病気の関係性に関する洞察を得られるようになると考えています。
サンプルデータの読み込み
サンプルの小規模コホート(集団)から変異データを読み込むところからスタートしましょう。以下の文では、特定のサンプルIDのデータを読み込み、Delta Lakeフォーマットで(delta_stream_outputフォルダに)保存しています。
spark.read. \
format("parquet"). \
load("dbfs:/annotations_etl_parquet/sampleId=" + "SRS000030_SRR709972"). \
write. \
format("delta"). \
save(delta_stream_outpath)
注意
annotations_etl_parquetフォルダはParquetフォーマットで保存された1000 genomes datasetから生成されたアノテーションを含んでいます。ETLおよびアノテーションの処理にはDatabricks’ Unified Analytics Platform for Genomicsが使用されています。
Delta Lakeテーブルによるストリーミングの開始
以下の文においては、Sparkデータフレームexomes(試験)を作成し、Delta Lakeフォーマットを使用して(readStreamを通じて)データのストリームを読み込みます。これは継続的に実行する、動的なデータフレームです。すなわち、 delta_stream_outputフォルダにデータが書き込まれるたびに、exomesデータフレームは新たなデータを読み込みます。exomesデータフレームを参照するには、sampleIdでグループされた変異体の数をカウントするクエリーをデータフレームに対して実行します。
# Read the stream of data
exomes = spark.readStream.format("delta").load(delta_stream_outpath)
# Display the data via DataFrame query
display(exomes.groupBy("sampleId").count().withColumnRenamed("count", "variants"))
display関数を実行すると、Databricksノートブックはストリーミングジョブをモニタリングするためのストリーミングダッシュボードを表示します。下の図がdisplay関数で表示されるストリーミングジョブです。sampleIdごとの変異体の数を表示しています。
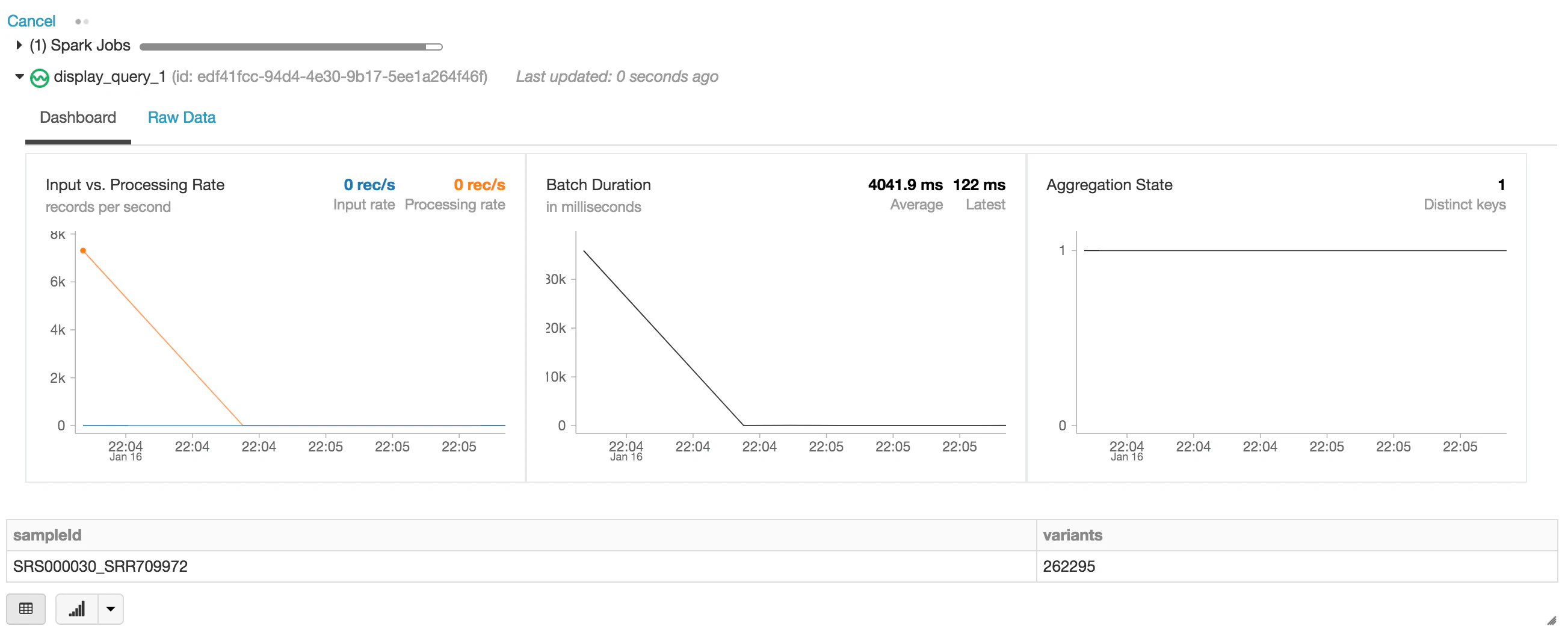
exomesデータフレームに対してクエリーを実行することで、最初に提示した質問への回答を進めましょう。
一塩基ヌクレオチド変異体のカウント
次に、一塩基ヌクレオチド変異体(Single Nucleotide Variant:SNV)の数を計算して以下のグラフを表示します。
%sql
select referenceAllele, alternateAllele, count(1) as GroupCount
from snvs
group by referenceAllele, alternateAllele
order by GroupCount desc

注意
displayコマンドは、コーディングなしにデータフレームの中身を可視化できるDatabricksの提供機能です。
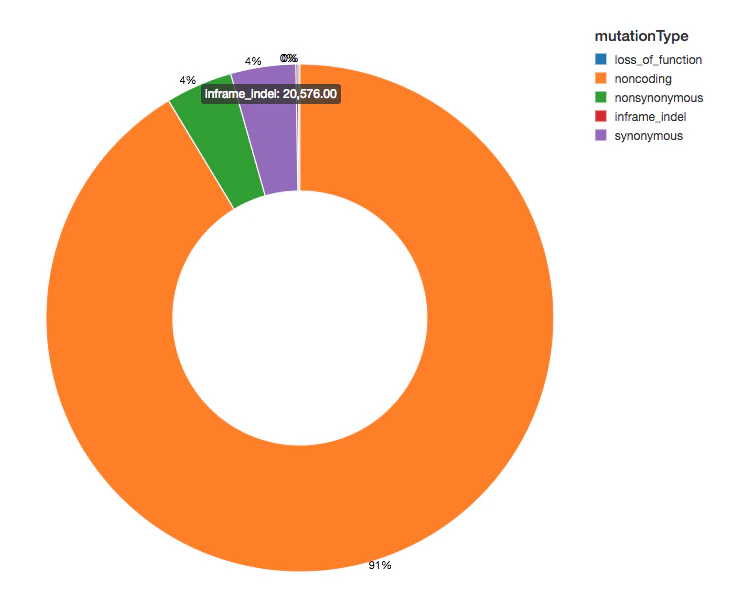
変異体のカウント
機能的影響に基づいて変異体にアノテーションを追加しているので、観測した変異体の影響の分布を確認することで分析を継続します。変異体の多くはタンパク質を示すフランキング領域を検知しており、これらはノンコーディング変異体と呼ばれます。
display(exomes.groupBy("mutationType").count())
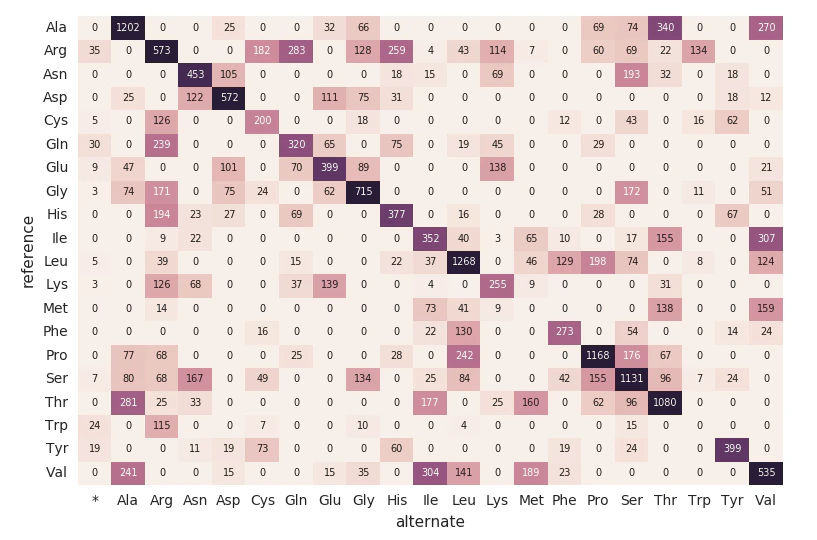
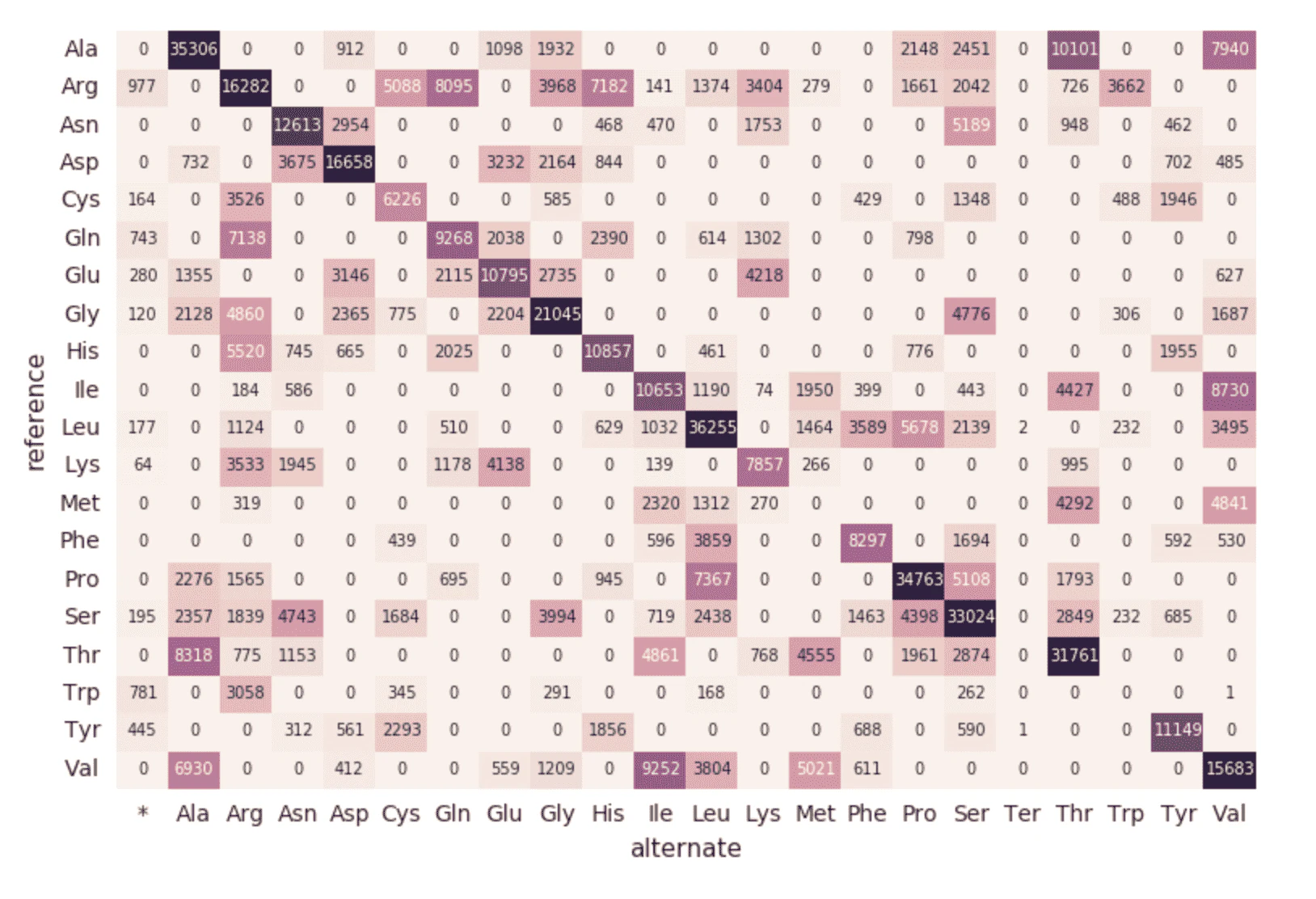
アミノ酸置換ヒートマップ
exomesデータフレームの分析を続けます。次に以下のコードを使用してアミノ酸置換(Amino Acid Substitution)をカウントしましょう。上のデータフレームと同様に、別の動的なデータフレーム(aa_counts)を作成し、exomesデータフレムによって新たデータが処理されるようにし、結果的にアミノ酸置換のカウントも更新されるようにします。また、ここではデータをメモリーに書き込み(format("memory"))、60秒間隔のバッチで処理(trigger(processingTime='60 seconds'))を行うことで、下流のpandasヒートマップのコードがヒートマップの処理、可視化を行えるようにします。
# Calculate amino acid substitution counts
coding = get_coding_mutations(exomes)
aa_substitutions = get_amino_acid_substitutions(coding.select("proteinHgvs"), "proteinHgvs")
aa_counts = count_amino_acid_substitution_combinations(aa_substitutions)
aa_counts. \
writeStream. \
format("memory"). \
queryName("amino_acid_substitutions"). \
outputMode("complete"). \
trigger(processingTime='60 seconds'). \
start()
以下のコードは上のSparkテーブルamino_acid_substitutionsを読み込み、カウントの最大値を特定し、Sparkテーブルから新たなpandasピボットテーブルを作成し、ヒートマップをプロットします。
# Use pandas and matplotlib to build heatmap
amino_acid_substitutions = spark.read.table("amino_acid_substitutions")
max_count = amino_acid_substitutions.agg(fx.max("substitutions")).collect()[0][0]
aa_counts_pd = amino_acid_substitutions.toPandas()
aa_counts_pd = pd.pivot_table(aa_counts_pd, values='substitutions', index=['reference'], columns=['alternate'], fill_value=0)
fig, ax = plt.subplots()
with sns.axes_style("white"):
ax = sns.heatmap(aa_counts_pd, vmax=max_count*0.4, cbar=False, annot=True, annot_kws={"size": 7}, fmt="d")
plt.tight_layout()
display(fig)
継続的パイプラインへの移行
ここまでにおいては、上記のコード、可視化は単一のsampleIdに対する単一の処理に対するものでした。しかし、我々はDatabricks Delta Lakeと構造化ストリーミングを使用しているので、(変更なしに)このコードを用いて、サンプルがパイプラインに投入されるのに合わせて継続的に品質管理メトリクスを計算するプロダクションデータパイプラインを構築することができます。これをデモンストレーションするために、データセット全体をロードする以下のコードを実行します。
import time
parquets = "dbfs:/databricks-datasets/genomics/annotations_etl_parquet/"
files = dbutils.fs.ls(parquets)
counter=0
for sample in files:
counter+=1
annotation_path = sample.path
sampleId = annotation_path.split("/")[4].split("=")[1]
variants = spark.read.format("parquet").load(str(annotation_path))
print("running " + sampleId)
if(sampleId != "SRS000030_SRR709972"):
variants.write.format("delta"). \
mode("append"). \
save(delta_stream_outpath)
time.sleep(10)
上述のコードで説明したように、exomesデータフレームはdelta_stream_output フォルダにロードされたファイルです。最初は単一のsampleId(sampleId = "SRS000030_SRR709972")のデータをロードしました。上のコードは生成されたすべてのParquetサンプルを取り込み、sampleIdごとにdelta_stream_outputフォルダにインクリメンタルにロードします。以下のアニメーションは上のコードのアウトプットを表示しています。
ゲノミクスパイプラインの可視化
ノートブックの先頭にスクロールすると、今やexomesは自動で新たなsampleIdを自動的に読み込んでいることに気づくかと思います。我々のゲノミクスパイプラインの構造化ストリーミングコンポーネントは継続的に実行されるため、新たなデータがdelta_stream_outputpathフォルダにロードされるたびにデータを処理します。Databricks Delta Lakeフォーマットを使うことで、exomesデータフレームに流れ込むストリーミングデータのトランザクション一貫性を保証します。
最初にexomesデータフレームを作成した時と異なり、ストリーミングダッシュボードでどのように新たデータがロードされているのか("input vs. processing rate"、"batch duration"の変動、"aggregations state"における別個のキーの数の増加)に注意してください。exomesデータフレームの処理が進むのに合わせて、新たなsampleId(および変異体の数)に注意してください。同様の動作は関連づけられている変異体タイプのクエリーでも確認できます。
DatabricksのDelta Lakeを活用することで、ゲノミクスパイプラインの各ステップにおける新規データのトランザクション一貫性は保持されます。このことは、あなたのパイプラインは一貫性があり(すべてのデータが「正しい」ことを保証します)、信頼性があり(トランザクションは成功するか失敗するかのどちらかです)、リアルタイムの更新を取り扱える(多くのトランザクションを同時に取り扱え、あらゆる障害もデータに影響を与えません)という点で重要と言えます。つまり、下流の(追加ETLステップを含む)アミノ酸置換のヒートマップもシームレスに更新されます。
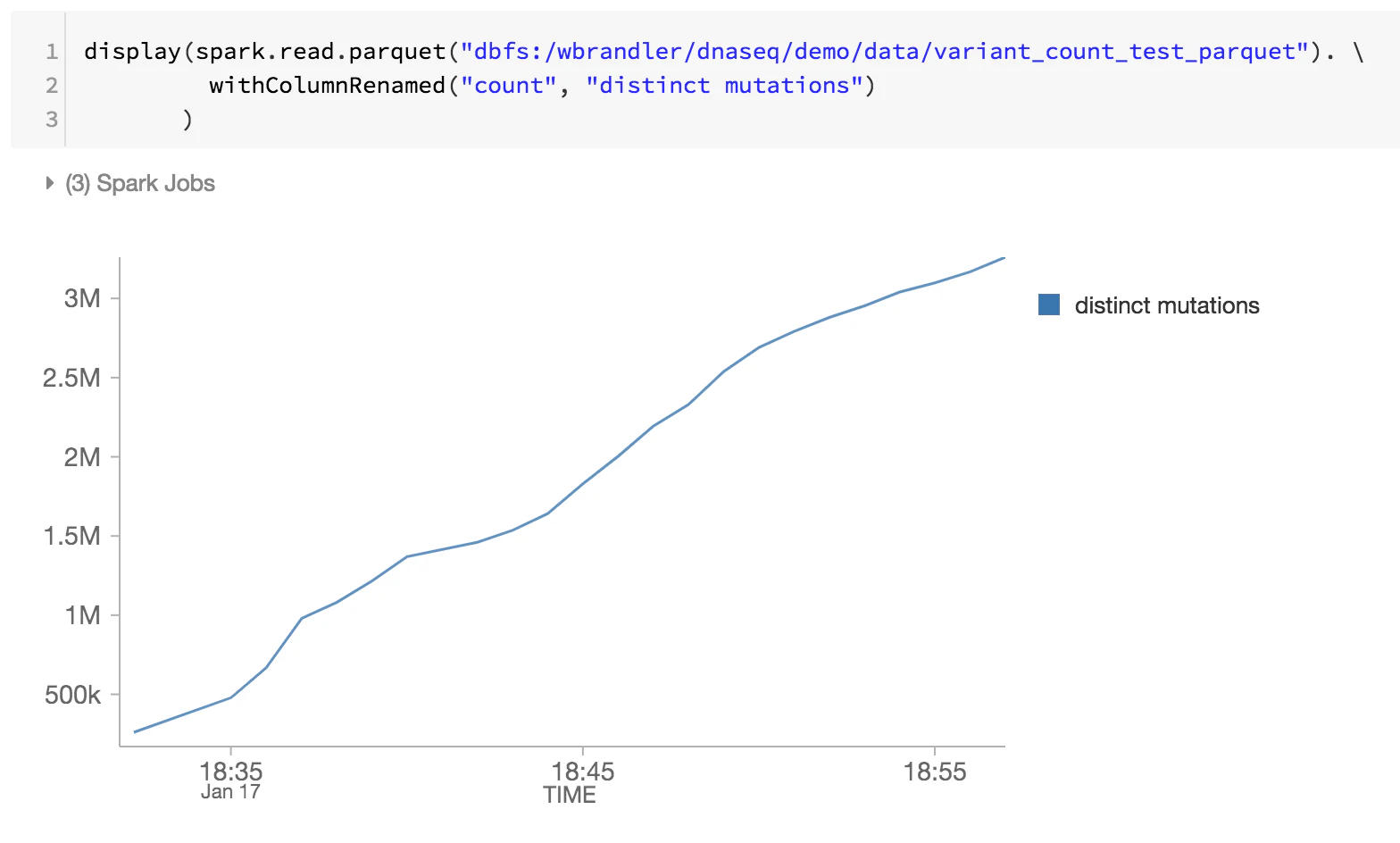
ゲノミクスパイプラインの最後のステップとして、DBFS内のDelta Lakeファイルを確認し、別個の変異体の数(時間経過に対する別個の変異体数の数の増加)をモニタリングします。
サマリー
Databricksレイクハウスプラットフォーム、特にDelta Lakeの基盤、そしてDatabricks Unified Analytics Platform for Genomicsを活用することで、バイオインフォマティクス専門家と研究者は、トランザクション一貫性を維持したまま分散処理による分析を行うことが可能となります。これらの抽象化によってデータ実践者はゲノミクスパイプラインを簡略化できます。ここでは、新たなサンプルが処理されるのに合わせて、人手の介在なしに継続的にデータを処理するゲノミクスサンプルの品質管理パイプラインを構築しました。ETLを行っていても、先進的な分析を行っていても、あなたのデータは高速かつ妨害を受けることなしにゲノミクスパイプラインを流れていきます。サンプルノートブックをダウンロードして試してみてください。
大規模ゲノミクスデータを分析するには以下を参考にしてください。
- ゲノミクス統合分析ソリューションガイドを一読ください。
- サンプルノートブックDelta Lakeによる大規模ゲノミクスパイプラインの簡略化をダウンロードしてください。
- Databricks 無料トライアルにサインアップしてください。
謝辞
Yongsheng Huang、Michael Ortegaに謝意を表します。
Databricks 無料トライアル
-
The Human Genome ProjectはDepartment of EnergyとNational Institutes of Health主導で1990年から2003年の間に進められた30億ドルのプロジェクトです。 ↩