Rを使ってゴリゴリ微生物群集の解析を行っていこうと思う。
群集の相違を可視化する際、PCA(主成分分析)やPCoA(主座標分析)などの手法もあるが、最近の勉強によると生物群集の構成の相違にはNMDS(非多次元尺度構成法)がかなり使われているので今回はNMDSにする。
##使用したデータ
・微生物(OTU)のカウントデータ
以前の記事で紹介した、QIIME2の公式チュートリアルのサンプルデータ(table.qza)からQIIME2を用いて抽出し、得た微生物群集のカウントデータを使って解析を行っていく。
その記事でも述べたが、邪魔なので1行目を削除し、1列目×2行目の"#OTU ID"を"ID"に変えて、ファイル名をotu_count.tsvとした。
※行が各サンプル、列が各OTUを表しているデータフレーム。
以前の記事のやり方でQIIME2から取ってくると行列が入れ替わって出てくるので、下のコマンドで逆にしている。
・メタデータ(環境要因のデータ)
QIIME2の公式チュートリアルのデータを使う。
このリンクで得られるデータの、2行目を消して、1行目の-(ハイフン)を_(アンダーバー)に変え、sample_metadata.tsvとして保存した。
なんとなく、ハイフンは「マイナス」の意味もあるのであまり使いたくはない、、、
※行が各サンプル、列が各環境要因を表しているデータフレーム
##veganで解析
読み込み作業
#veganパッケージの読み込み
library(vegan)
#カウントデータの読み込み
community <- read.table("otu_count.tsv", header = T, row.names = 1)
#行名がサンプル名、列名がOTU名のほうが何かとやりやすいので行列の入れ替え
community <- as.data.frame(t(community))
#メタデータの読み込み
env <- read.table("sample_metadata.tsv", header = T)
QIIME2はカウントの絶対値が出てくる。
カウントの絶対値の合計はサンプルによって違う(シークエンシングの際に読めたリード数にも依存している)ので、それらを比較しても不公平である。
そこですべてのサンプルから同じ数だけランダムにカウントを取ってくる、rarefactionという作業を行う。
ランダムに取ってくる数は、サンプル内の合計カウントが最小のサンプルに合わせる(そのサンプルは最大でもそれだけしか取れないので)
rarefaction
#行の合計が最小のものでrarefaction
rared_com <- rrarefy(community, min(rowSums(community)))
NMDSには、metaMDS関数を使う。
#NMDS
ord <- metaMDS(rared_com)
正直、veganを使ったNMDSの可視化は色んな人がやっているので、普通のやり方で十分な人は、他のサイトを見ていいと思う。
例えば、このサイト
私はあくまでggplot2を使って、このNMDSデータを可視化したかった。
なぜならggplotに慣れている人は、ggplotのほうが線の太さ等を変える方法がわかりやすいと思うからである。
てことで普通のveganでできるNMDSプロットは飛ばして、このordをggplot2で使えるような形にしていきたい。
##veganデータの取り出し
ggplot系では、サンプル名の列と、それに対応した他の変数の列がいくつか並んだ形のデータフレームでないと図を作れない。
それを作っていく。
ある列の値をもとに結合するときはmerge関数を使う。
#scores関数でordのNMDS1,2の値を取り出す
data.scores <- as.data.frame(scores(ord))
#列(sample_id)を作ってそこに行名を入れる
#次でenvと結合するために、あえてenv中のサンプル名のヘッダーと同名にした
data.scores$sample_id <- rownames(data.scores)
#メタデータを結合してしまう。byで、両データにあるsample_idによってきちんと対応させる。
data.scores <- merge(data.scores, env, by="sample_id", sort=F)
#確認
head(data.scores)
以下のような、NMDS1、NMDS2の値と、メタデータがsample_idに従って並べられたデータフレームができる。
| sample_id | NMDS1 | NMDS2 | ・・・ | year | ・・・ |
|---|---|---|---|---|---|
| L1S105 | -2.319019 | 0.33685618 | ・・・ | 2009 | ・・・ |
| L1S140 | -2.436710 | -0.06625757 | ・・・ | 2008 | ・・・ |
| L1S208 | -2.547093 | 0.17384921 | ・・・ | 2009 | ・・・ |
| L1S257 | -2.531821 | 0.08251122 | ・・・ | 2009 | ・・・ |
| L1S281 | -2.738402 | 0.25667777 | ・・・ | 2009 | ・・・ |
| : | : | : | : | : |
##ggplot2でプロットする
点でのプロット
被験者(subject)を色で、体の部位(bodt_site)を点の形で表すことにした。
因子が2つない場合は、形(shape)か色(colour)のどちらかを指定しなければいい。
#パッケージの読み込み
library(ggplot2)
#点だけのプロットの作成
g <- ggplot() +
geom_point(data=data.scores, aes(x=NMDS1, y=NMDS2, shape=body_site, colour=subject), size=8) +
scale_colour_manual(values=c("subject-1"="red", "subject-2"="blue")) + #色を指定
scale_shape_manual(values = c("gut"=16, "left palm"=15, "right palm"=17, "tongue"=18)) + #形を指定
theme_bw() +
theme(panel.border = element_rect(size = 1.8, colour = "black"),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank(),
legend.position = "right",
legend.background = element_rect(fill = "white", colour = "black"), #凡例の背景や色
legend.title = element_text(size = 25, face = "bold"), #凡例タイトルや中身のテキストの大きさや字体の調整
legend.text = element_text(size = 25, face = "bold"),
axis.text=element_text(size = 25, face = "bold", colour = "black"),
axis.title=element_text(size = 35,face = "bold", colour = "black")) +
labs(shape="Body site", colour="Subject") #凡例の名前をきちんとする
g
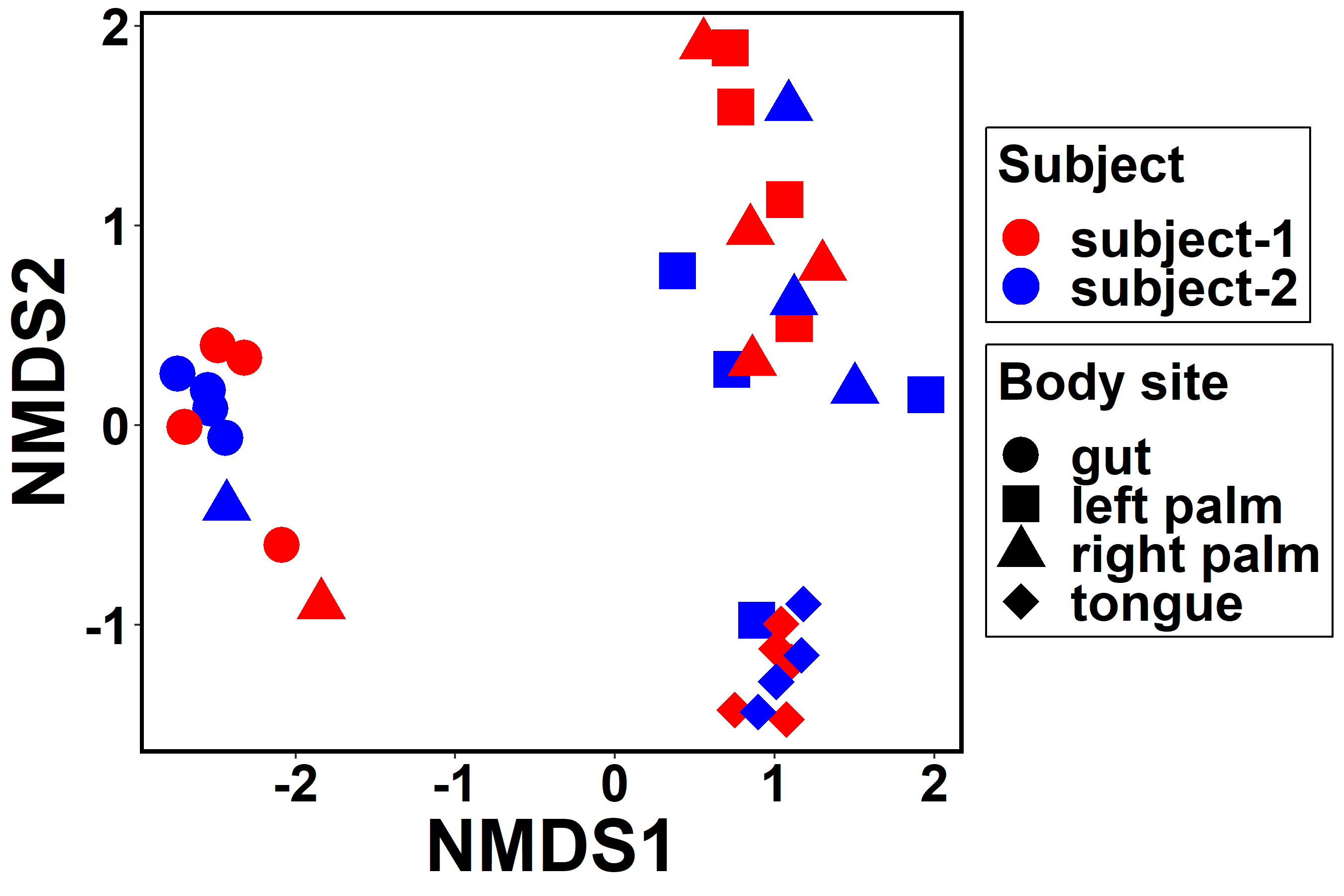
点どうしの距離が、そのサンプルどうしの群集構造の違いを表しています。
微生物の群集構造は、被験者による違いより部位による違いのほうが大きそう。
##polygonにする
論文とかを見ていると、群ごとにプロットの外周を囲う枠が見れると思う。
あれがpolygonだと個人的に解釈している。
veganならordispiderとかでできるのだが、今回はggplot2でやりきると決めた手前、ggplot2と心中する。
##1因子でのpolygon
これまでは被験者(subject)と体の部位(body_site)の2因子を用いてきたが、簡易にするために、まずは因子を1つ減らしたものを使ってみる(いきなり2因子は説明することが多すぎる)。
ということで、被験者の違いはいったん無視してdata.scores内の、体の部位を色で指定して作ることにする。
点の形は指定せずに色だけ指定して作るとこうなる。
g1 <- ggplot() +
geom_point(data=data.scores, aes(x=NMDS1, y=NMDS2, colour=body_site), size=8) +
scale_colour_manual(values=c("gut"="red", "left palm"="blue", "right palm"="darkgreen", "tongue"="orange")) +
theme_bw() +
theme(panel.border = element_rect(size = 1.8, colour = "black"),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank(),
legend.position = "right",
legend.background = element_rect(fill = "white", colour = "black"), #凡例の背景や色
legend.title = element_text(size = 25, face = "bold"), #凡例タイトルや中身のテキストの大きさや字体の調整
legend.text = element_text(size = 25, face = "bold"),
axis.text=element_text(size=25, face = "bold", colour = "black"),
axis.title=element_text(size=35,face="bold", colour = "black")) +
labs(colour="Body site")
g1 #確認
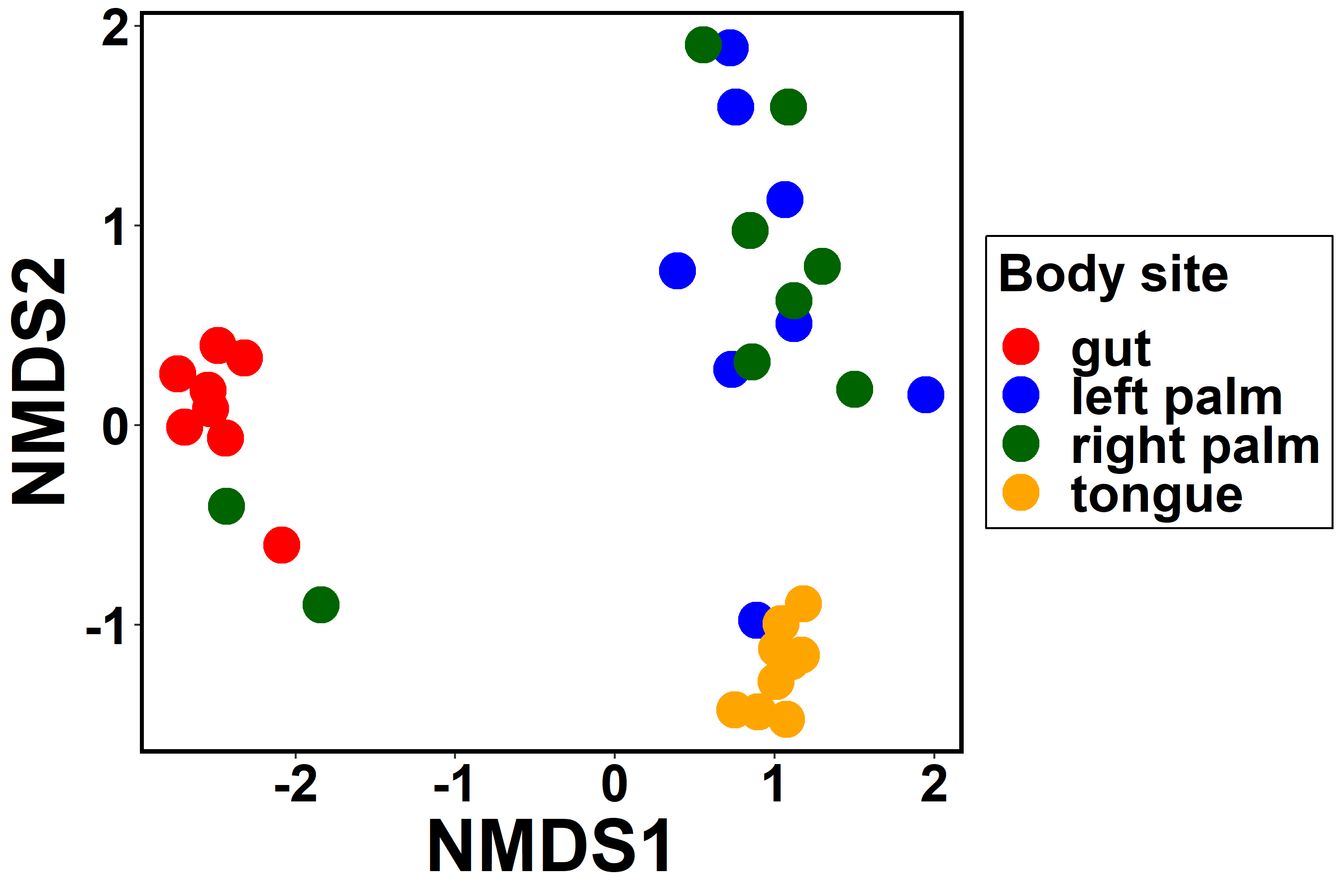
続いて、polygonにするには、この各群の点の外側を囲う、すなわち外側にある点のみのデータを作らないといけない。
これはchullという関数で行う。
#gutの点の集まりのうち、外側のもののNMDS1,NMDS2を取得。以下同様。
gut_hull <- data.scores[data.scores$body_site == "gut", ][chull(data.scores[data.scores$body_site == "gut", c("NMDS1", "NMDS2")]), ]
lp_hull <- data.scores[data.scores$body_site == "left palm", ][chull(data.scores[data.scores$body_site == "left palm", c("NMDS1", "NMDS2")]), ]
rp_hull <- data.scores[data.scores$body_site == "right palm", ][chull(data.scores[data.scores$body_site == "right palm", c("NMDS1", "NMDS2")]), ]
ton_hull <- data.scores[data.scores$body_site == "tongue", ][chull(data.scores[data.scores$body_site == "tongue", c("NMDS1", "NMDS2")]), ]
hullったデータをggplotで使う用にがっちゃんこする。
データフレームの縦の結合はrbindでできる。
hulldata <- rbind(gut_hull, lp_hull, rp_hull, ton_hull)
hulldataをgeom_polygonに入れる
すっきりさせるためにshow.legend=Fがけっこうポイント。
#g1に上書きした
g1 <- g1 +
geom_polygon(data=hulldata,
aes(x=NMDS1,y=NMDS2,fill=body_site),
alpha=0.3,
#ここでshow.legend = Fとしないと、polygonのための凡例がでてくるので消しておく。
show.legend = F) +
scale_fill_manual(values=c("gut"="red", "left palm"="blue", "right palm"="darkgreen", "tongue"="orange"))
g1 #確認
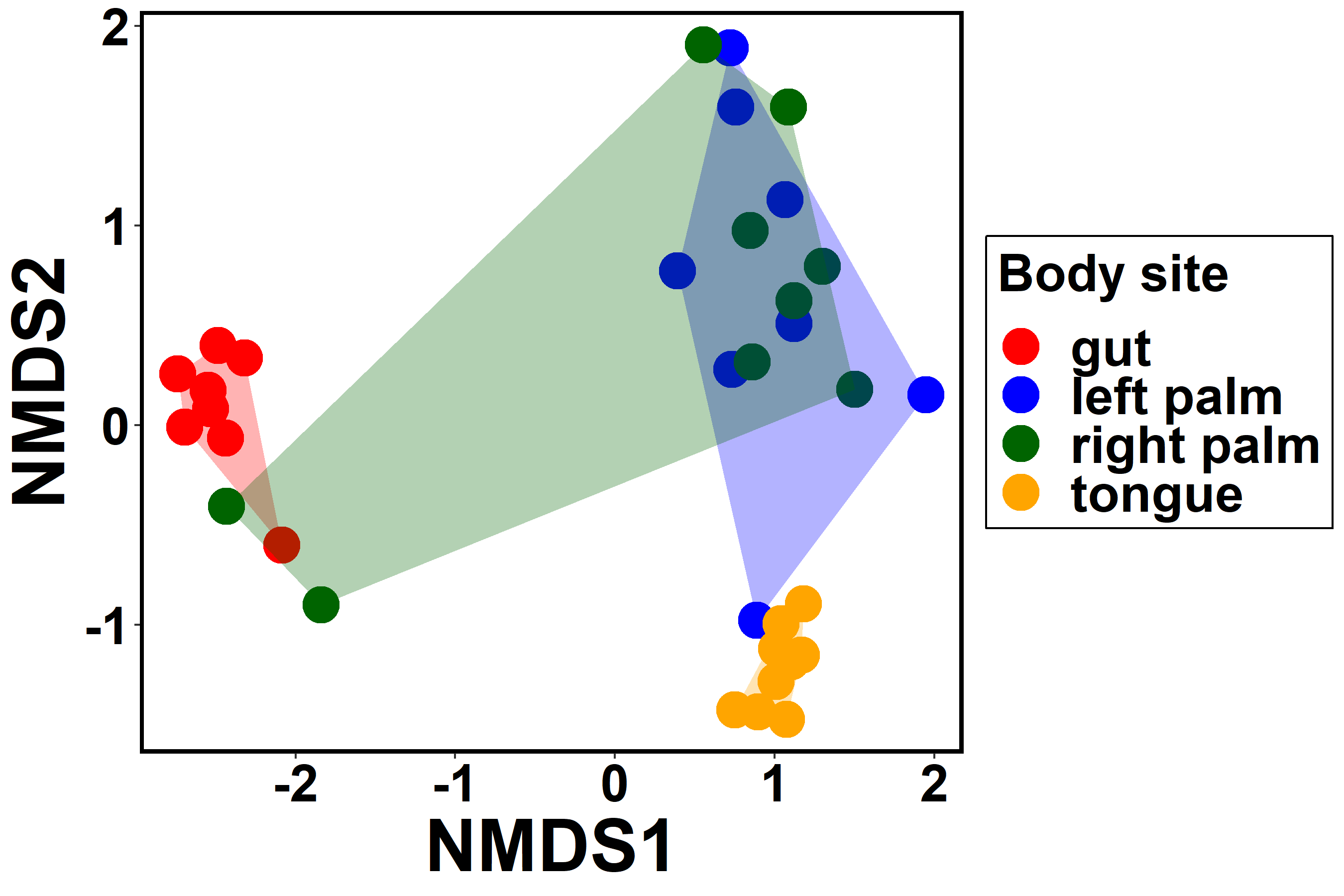
これで、体の各部位の微生物コミュニティーがどれぐらい互いの部位と違ったり似ているか少しわかりやすくなったと思う。
##応用編:2因子でのpolygon
2因子では、同じ色のものを全て1くくりに囲ってしまっては、もう1つの因子の効果が見づらくなる。
そこで今回なら、被験者(subject)×体の部位(body_site)という見方をする必要があり、2被験者×4部位で8通りのグループができることになる。
subjectの列とbody_siteの列の文字列を結合させた新たな列を作ることができれば、8通りのパターンを作ることができる。
dplyrパッケージのmutate関数で列の追加。multiという列を追加した。
pasteでbody_site列とsubject列の文字列を_によって結合する。それがmultiに入る。
library(dplyr)
data.scores2 <- data.scores %>%
mutate(multi = paste(!!!rlang::syms(c("body_site", "subject")), sep = "_"))
# 追記2023.01.20.:rlangがうまく動かなくなったが、mutate(multi = paste(body_site, "_", subject))などで代替できた。
次は今回で最もしんどいところだと思う。
8通りのパターンのhullデータ(上述)を作り、rbindで結合する。
gut_hull1 <- data.scores2[data.scores2$multi == "gut_subject-1", ][chull(data.scores2[data.scores2$multi == "gut_subject-1", c("NMDS1", "NMDS2")]), ]
gut_hull2 <- data.scores2[data.scores2$multi == "gut_subject-2", ][chull(data.scores2[data.scores2$multi == "gut_subject-2", c("NMDS1", "NMDS2")]), ]
lp_hull1 <- data.scores2[data.scores2$multi == "left palm_subject-1", ][chull(data.scores2[data.scores2$multi == "left palm_subject-1", c("NMDS1", "NMDS2")]), ]
lp_hull2 <- data.scores2[data.scores2$multi == "left palm_subject-2", ][chull(data.scores2[data.scores2$multi == "left palm_subject-2", c("NMDS1", "NMDS2")]), ]
rp_hull1 <- data.scores2[data.scores2$multi == "right palm_subject-1", ][chull(data.scores2[data.scores2$multi == "right palm_subject-1", c("NMDS1", "NMDS2")]), ]
rp_hull2 <- data.scores2[data.scores2$multi == "right palm_subject-2", ][chull(data.scores2[data.scores2$multi == "right palm_subject-2", c("NMDS1", "NMDS2")]), ]
ton_hull1 <- data.scores2[data.scores2$multi == "tongue_subject-1", ][chull(data.scores2[data.scores2$multi == "tongue_subject-1", c("NMDS1", "NMDS2")]), ]
ton_hull2 <- data.scores2[data.scores2$multi == "tongue_subject-2", ][chull(data.scores2[data.scores2$multi == "tongue_subject-2", c("NMDS1", "NMDS2")]), ]
hulldata_multi <- rbind(gut_hull1, gut_hull2, lp_hull1, lp_hull2, rp_hull1, rp_hull2, ton_hull1, ton_hull2)
ようやくプロットに移る。
実は2因子のプロットは1番上でやっているので、gがまだ使える。そこにpolygonをするためのhulldata_multiを入れる。
fill(塗りつぶし)はsubject、
group(どうくくるか)はmultiにするのがポイント。
こうすることで、色は同じでも、さっきの8パターンの点でくくってくれる。
gm <- g +
geom_polygon(data=hulldata_multi,
aes(x=NMDS1,y=NMDS2,fill=subject, group=multi),
alpha=0.3,
#ここでshow.legend = Fとしないと、polygonのための凡例がでてくるので消しておく。
show.legend = F) +
scale_fill_manual(values=c("subject-1"="red", "subject-2"="blue"))
gm #確認
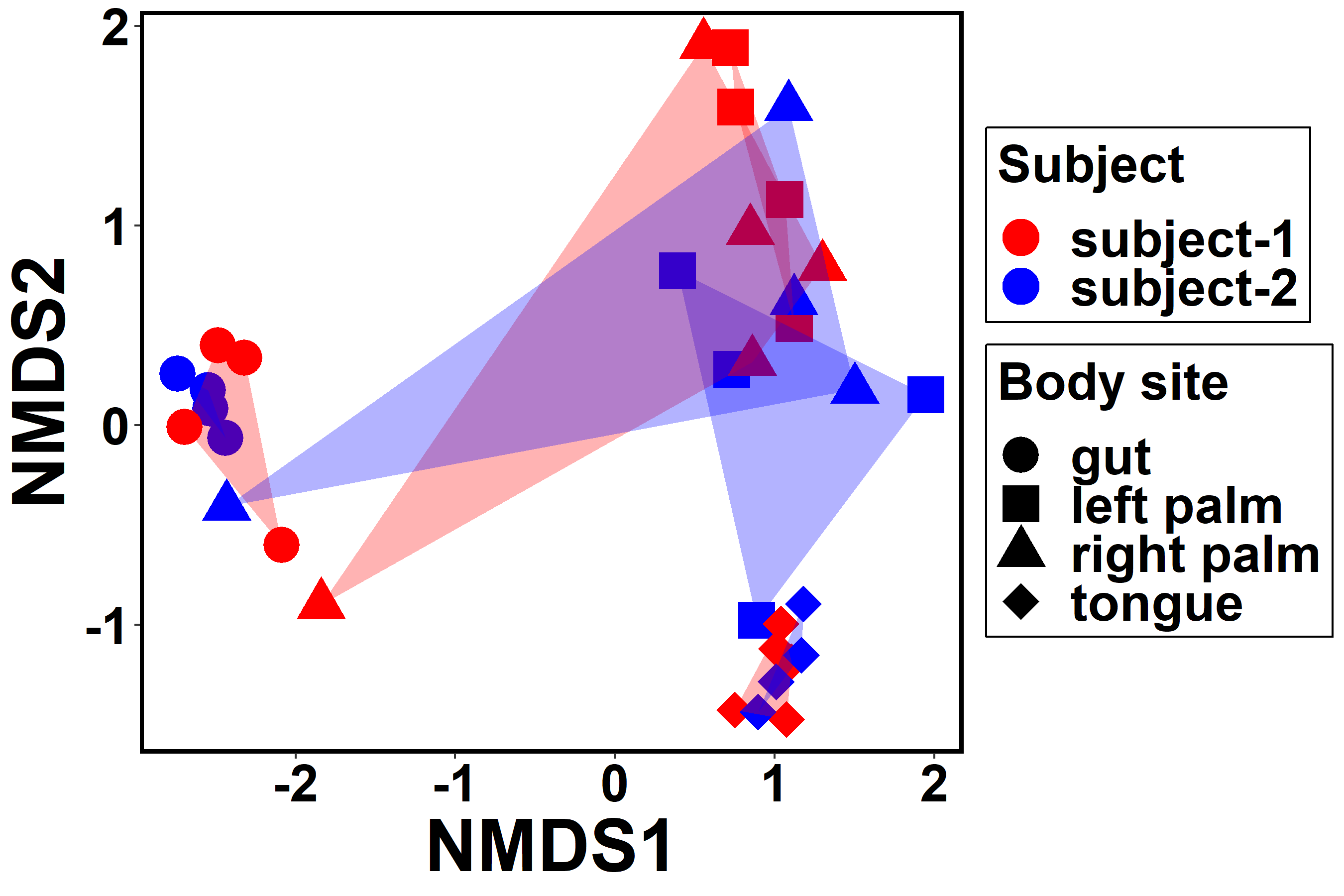
データの反復が少なめなので、外側にくるのが3点か4点だからけっこうカクカクしている笑
反復が増えればもうちょっとカッコよくなると思う。
##画像の保存
ggsave(filename = "作る画像名.png", plot = プロット名, width = 9, height = 6, dpi = 300)
触れていなかったが、RStudio上では画像が実際どんな感じかわからないので、適宜保存したほうがいいと思う。
プロット名というのは、ggplot2でつくった、上で言うg, g1, gmのことである。
##参考にさせていただいたサイト
NMDSについて
https://yokazaki.hatenablog.com/entry/2016/06/29/212153
plotについて
https://chrischizinski.github.io/rstats/vegan-ggplot2/