本書は抄訳であり内容の正確性を保証するものではありません。正確な内容に関しては原文を参照ください。
2000年代後半におけるゲノムワイド関連解析(GWAS)の到来により、研究者が糖尿病やクローン病などの多因子疾患の原因について最も根本的なレベルで理解できることができるようになりました。しかしながら、GWAS解析のための学術的なバイオインフォマティクスツールは、7ヶ月ごとに世界のデータ量が倍増するというゲノムデータの増加速度に追いついていけていません。
この大きな課題、そしてヘルスケアの将来に大きな意味を持つゲノミクスの重要性を踏まえ、Databricksでは専属エンジニアリングチームを結成し、高性能なビッグデータストアである Delta Lake や実行のログを記録するための MLflow を活用したGWASなどのためのSparkネイティブなワークフローの開発に従事させることとしました。これら3つの技術に私たちが社内開発したライブラリを組み合わせることで、集団レベルのゲノムデータに取り込む顧客が直面する課題を解決することが可能になります。
このツールには、bed、VCF、BGEN等のフォーマットのファイルをDelta Lakeに格納で共有した、ゲノムデータのフラットファイルを直接取り込むようなアーキテクチャも含まれます。このブログで私たちは共通の関連解析のツールをSpark SQLに移植し、ゲノムワイドの線形回帰のような共通の解析を効率化することに専念しました。次のブログでは、GWASツールSAIGEを始めとして、シングルノードのあらゆるバイオインフォマティクスツールを並列化するパイプライン処理を作成し、このプロセスを一般化したことについて説明します。
ここで、一般公開されている1000人ゲノムのデータセットを、1つのノートブックを使いどのように端から端までGWASワークフローを実行したかを紹介し、その結果を図1に示します。私たちはGWASカタログからの関連変異を用いてBMI(ボディマス指数)を人工的に生成しました(1000人ゲノムプロジェクトは表現型を含んでいないからです)。このノートブックはPythonで書かれていますが、R、Scala、SQLで実装することも可能です。
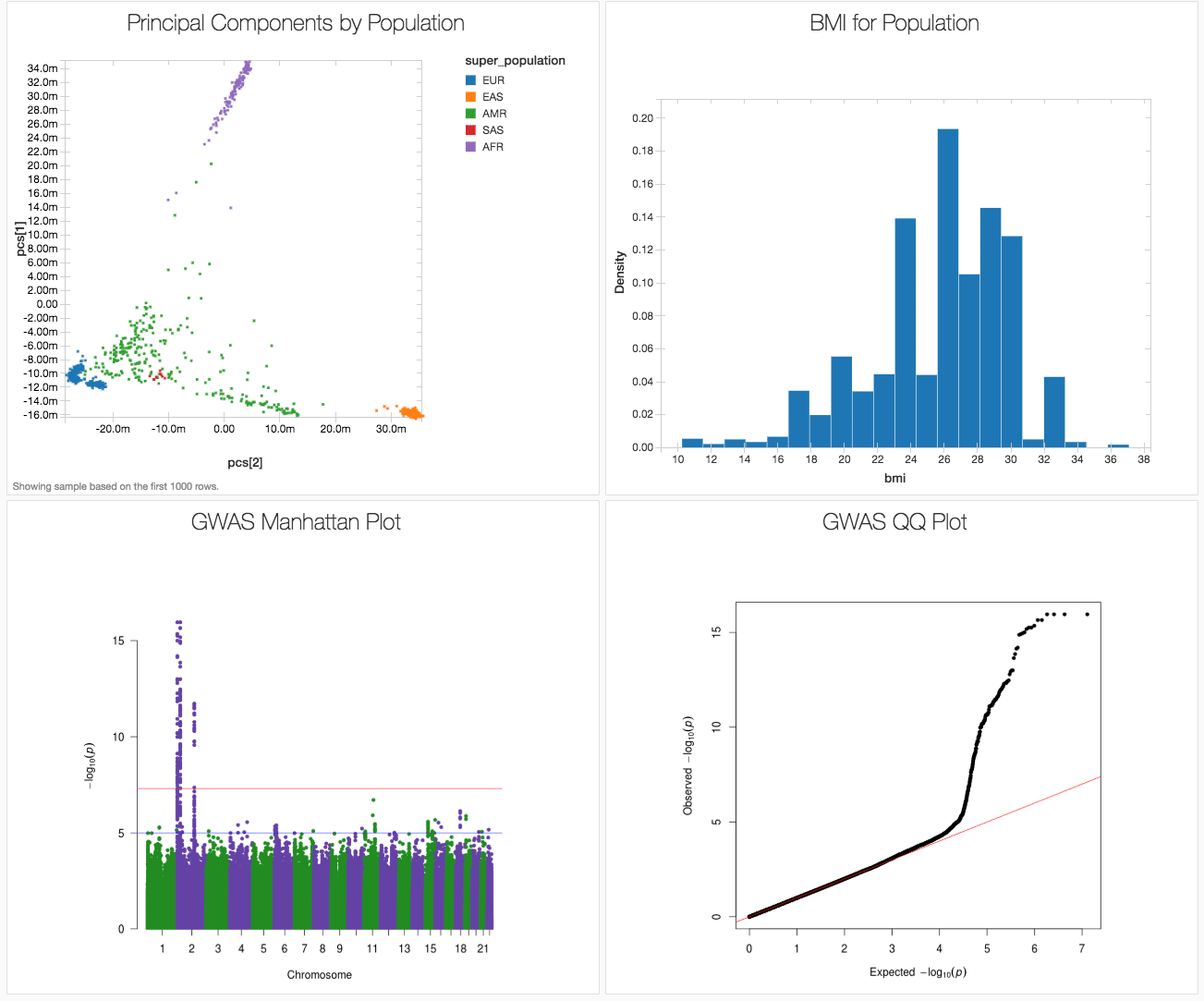
図1. Databricksダッシュボードで、1000人ゲノムデータセットに基づくシミュレーションデータを用いたGWASから得られた主要な結果を表示
1000人ゲノムのデータをDelta Lakeに取り込む
まず、1000人ゲノムのVCFファイルをSpark SQLのデータフレームとして読み込み、概要を示す統計情報を集計します。私たちのスキーマはゲノムの変異を表現する直観的なものであり、VCFやBGENを問わず一貫しています。
# VCFファイルをDatabricks Deltaバージョンとして読み込む
spark.read.format("com.databricks.vcf"). \
option("splitToBiallelic", "true"). \
option("flattenInfoFields", "false"). \
load(vcf_path). \
selectExpr("*", "expand_struct(call_summary_stats(genotypes))", "expand_struct(hardy_weinberg(genotypes))"). \
write. \
format("delta"). \
save(delta_path)
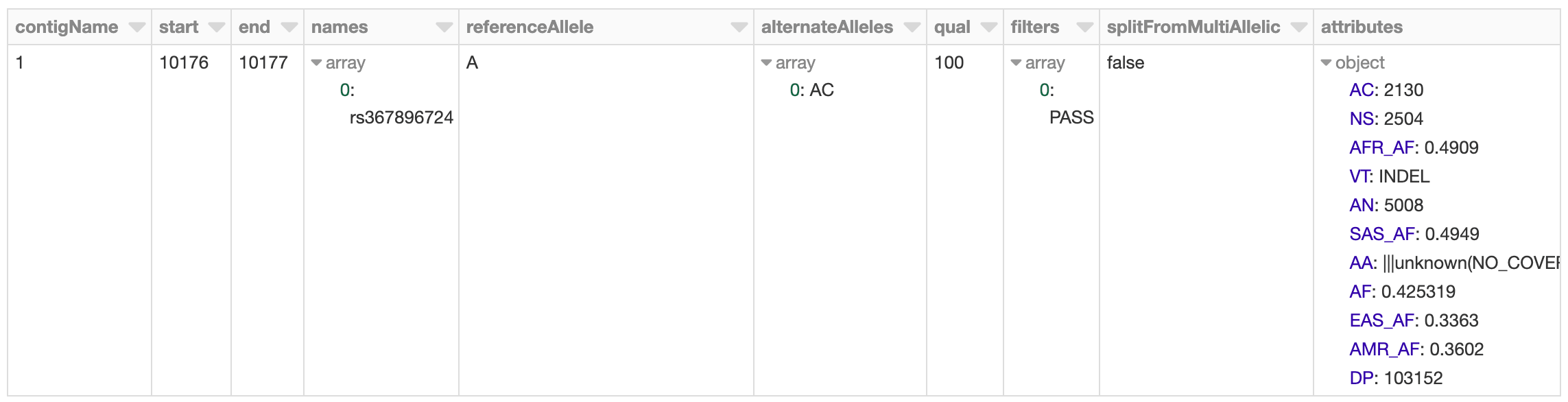
図2. Sparkデータフレームに格納されたVCFファイルをDatabricksの display() コマンドで表示したもの
1000人ゲノムのデータセットは全ゲノムシーケンスデータもあるため、多数の希少変異を含んでいます。データセットに対して集計のためのクエリーを実行したところ、8千万以上の変異があることがわかりました。それでは、このメトリクスをMLflowに記録してみましょう。
# VCFの合計数を計算
num_variants = spark.read.format("delta").load(delta_path).count()
mlflow.log_metric("Number Variants pre-QC", num_variants)
num_variants
# 出力結果
81271745
品質管理
私たちのゲノミクスライブラリには、単一変異についての遺伝子型について一般的に用いられる統計値を出力する関数や、単一サンプルについての統計値を全サンプルについて出力する関数などを含みます。ここで、ハーディー・ワインベルグ平衡(“pValueHwe”)を満たさない変異を除外してみます。ハーディー・ワインベルグ平衡は、集団遺伝学の統計値であり、正しく遺伝子型を検出できているかを検定するために用いられるものです。希少変異をアリル頻度を基準にして除外します。
spark.read.format("delta"). \
load(delta_path). \
where((col("alleleFrequencies").getItem(0) >= allele_freq_cutoff) &
(col("alleleFrequencies").getItem(0) <= (1.0 - allele_freq_cutoff)) & (col("pValueHwe") >= hwe_cutoff)). \
write. \
format("delta"). \
save(delta_qc_path)
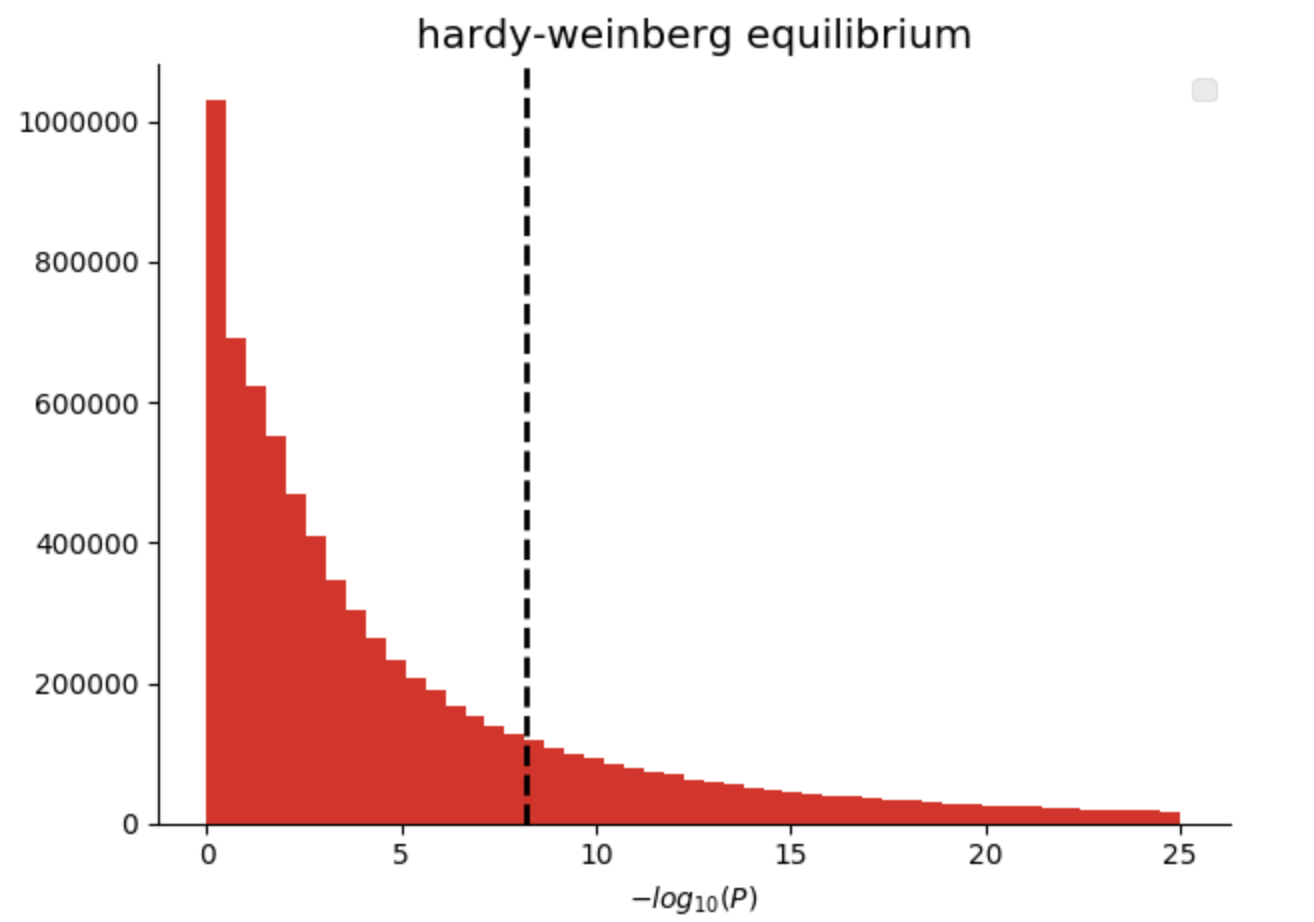
図3. ハーディー・ワインベルグ平衡のp値のヒストグラム
祖先型の把握
集団構造により遺伝子型と表現型の関連の解析を進めることができます。本研究の被験者間で異なる祖先型を把握するため、カーネル回帰の共変量として与えられる主成分(PC, Principal Components)を計算します。Sparkは、Spark MLLib DistributedMatrix APIを用いた特異値分解(SVD, Singular Value Decomposition)をサポートしており、遺伝子型行列の転置から主成分を出力するためにSVDを用いることができます。データフレームからDistributedMatrixを得ることを容易にするためにSparkのAPIを用い、それによりSVDを実行することで主成分を得ることができました。
vectorized = spark.read.format("delta"). \
load(delta_qc_path). \
selectExpr("array_to_sparse_vector(genotype_states(genotypes)) as features").cache()
matrix = RowMatrix(MLUtils.convertVectorColumnsFromML(vectorized, "features").rdd.map(lambda x: x.features))
pcs = matrix.computeSVD(num_pcs)
pcs_df = spark.createDataFrame(pcs.V.toArray().tolist(), ["pc" + str(i) for i in range(num_pcs)])
主成分分析を行った後にサンプルごとの主成分の密行列を得て、回帰分析の共変量に使いました。この後にするのは、サンプルID と 主成分 を抽出することです。これにより、1000人ゲノムのメタデータファイルに上位集団のラベルをつけることができました。
以下の散布図は、Databricksのdisplay()コマンドにより主成分のクラスターを表示したものです。
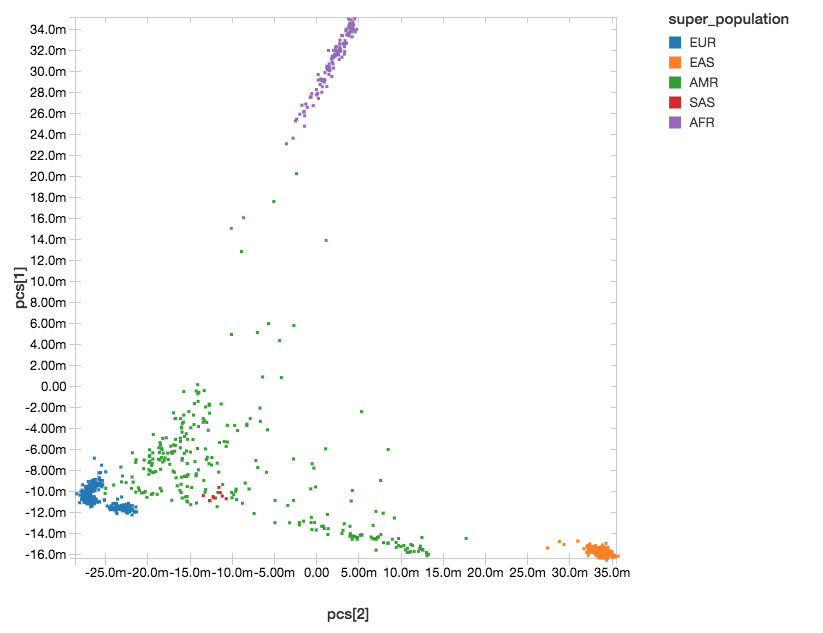
図4. 主成分分析の結果に上位集団のラベルをつけたもの
EUR = ヨーロッパ、EAS = 東アジア、AMR = 混合アメリカ、SAS = 南アジア、AFR = アフリカ
表現型データの取り込み
このゲノムワイド関連解析では、表現型としてBMIシミュレーションデータを用い、遺伝子型と関連づけました。遺伝子型データの取り込みと同様に、BMIデータもParquet形式データを読み込みます。
# 正規化された表現型データの取り込み
phenotypes_path = "dbfs:/databricks-datasets/genomics/1000G/phenotypes.normalized"
bmiPhenotype = spark.read. \
format("parquet"). \
load(phenotype_path). \
withColumnRenamed("values", "phenotype_values")
# BMIデータの表示
display(bmiPhenotype.selectExpr("explode(phenotype_values) AS bmi"))
上のdisplay()コマンドによりBMIのヒストグラムを表示できます
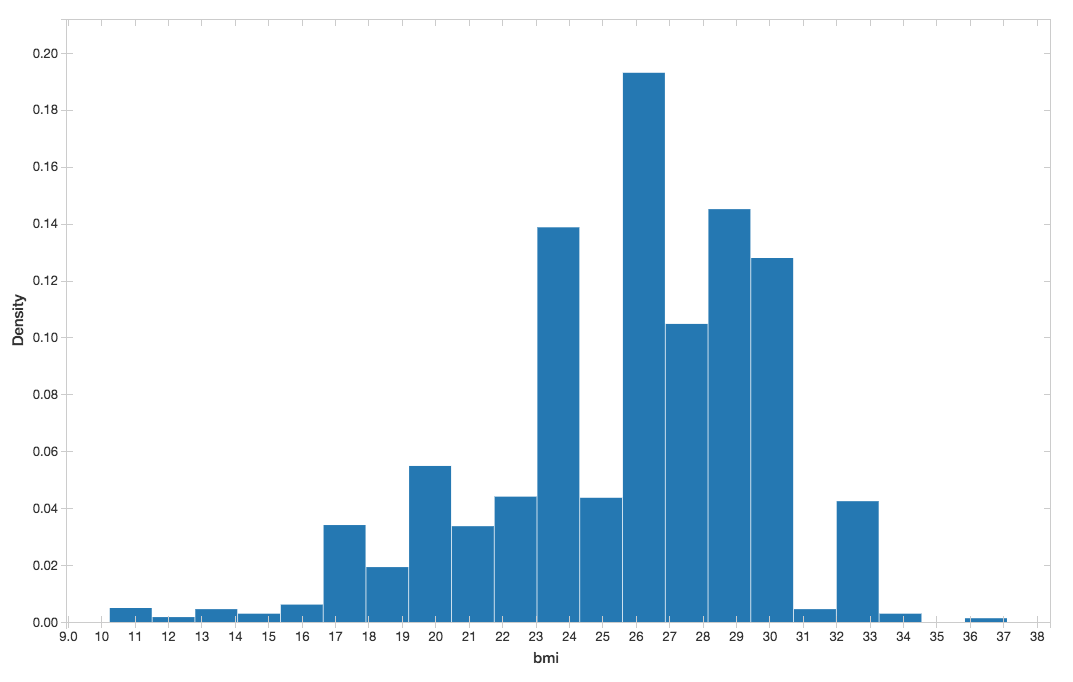
図5. BMIのヒストグラム
ゲノムワイド関連解析の実行
ここまでに、品質管理、データ抽出、変換とロード(ETL)など必要な処理を施してきました。ソリューションの次の手順は、以下のタスクによりGWASを実行することです。
- (
crossJoinを用いて)遺伝子型、表現型そして主成分をひもづけます - 線形回帰によりGWAS統計値を算出します
- GWAS統計値を含むApache Sparkのデータフレーム(
gwas_df)を構築します
# 遺伝子型、表現型そして主成分のクロスジョインにより変異をGWASへと関連させます
covariates = spark.read.format("delta").load(principal_components_path)
phenotypeAndCovariates = bmiPhenotype.crossJoin(covariates)
genotypes = spark.read.format("delta").load(delta_qc_path)
genotypes.crossJoin(phenotypeAndCovariates). \
selectExpr("contigName", "start", "phenotype", \
"expand_struct(linear_regression_gwas(genotype_states(genotypes), phenotype_values, covariates))"). \
write. \
format("delta"). \
save(gwas_results_path)
# データの表示
display(spark.read.format("delta").load(gwas_results_path))
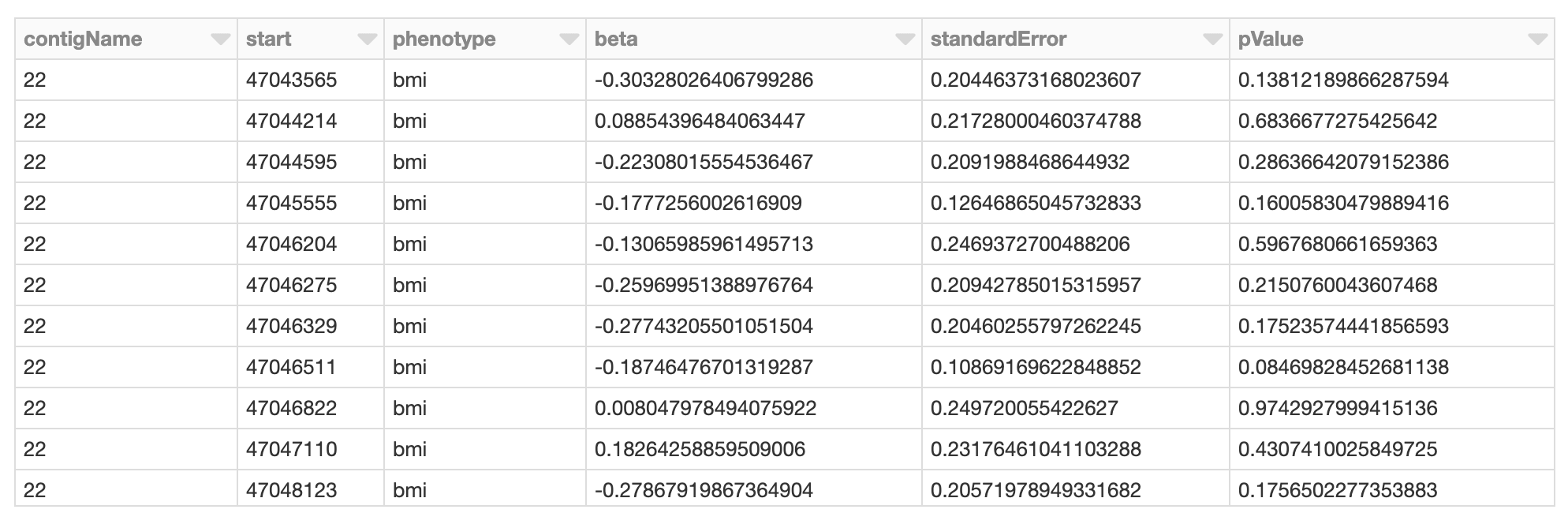
図6. GWASの結果を表すSparkデータフレーム
display()コマンドにより結果の吟味が可能となります。次に、qqmanパッケージを用いたゲノム横断的なマンハッタンプロットによる結果の可視化のため、PySparkのデータフレームをRに変換します。
# GWASの結果の抽出(そしてカラム名を新設)
gwas_results <- select(gwas_df, c(cast(alias(gwas_df$contigName, "CHR"), "double"), alias(gwas_df$start, "BP"), "P"))
# SparkデータフレームからRデータフレームへの変換
gwas_results_rdf <- as.data.frame(gwas_results)
# マンハッタンプロットのためのパッケージのインストール
install.packages("qqman", repos="http://cran.us.r-project.org")
library(qqman)
# GWASの結果のマンハッタンプロットとしての出力と、MLflowへの記録
png('/databricks/driver/manhattan.png')
manhattan(gwas_results_rdf,
col = c("#228b22", "#6441A5"),
chrlabs = NULL,
suggestiveline = -log10(1e-05),
genomewideline = -log10(5e-08),
highlight = NULL,
logp = TRUE,
annotatePval = NULL,
ylim=c(0,17))
dev.off()
mlflow.log_artifact('/databricks/driver/manhattan.png')
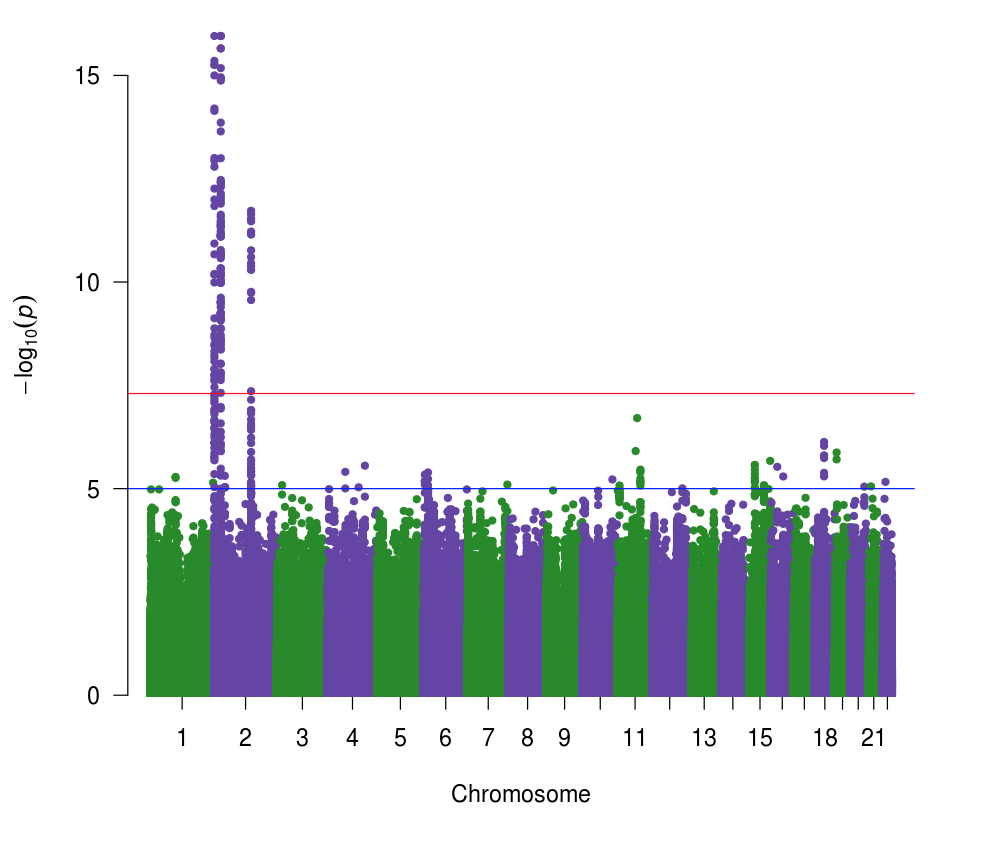
図7. GWASマンハッタンプロット
1000人ゲノムのシミュレーションデータを用いたゲノムワイド関連解析の結果からわかる通り、BMIに関連するいくつかの遺伝子座が2番染色体に見出されました。実際には、BMIに関連していることが判明している遺伝子座を、シミュレーションによるBMI表現型データ作成に使いました。
# QQプロット作成
qq(gwas_results_rdf$P)
Q-Qプロットを作図することで、正しく祖先を把握できたかを評価することができます。このケースでは、期待値からの偏りが真の関連を示しています。
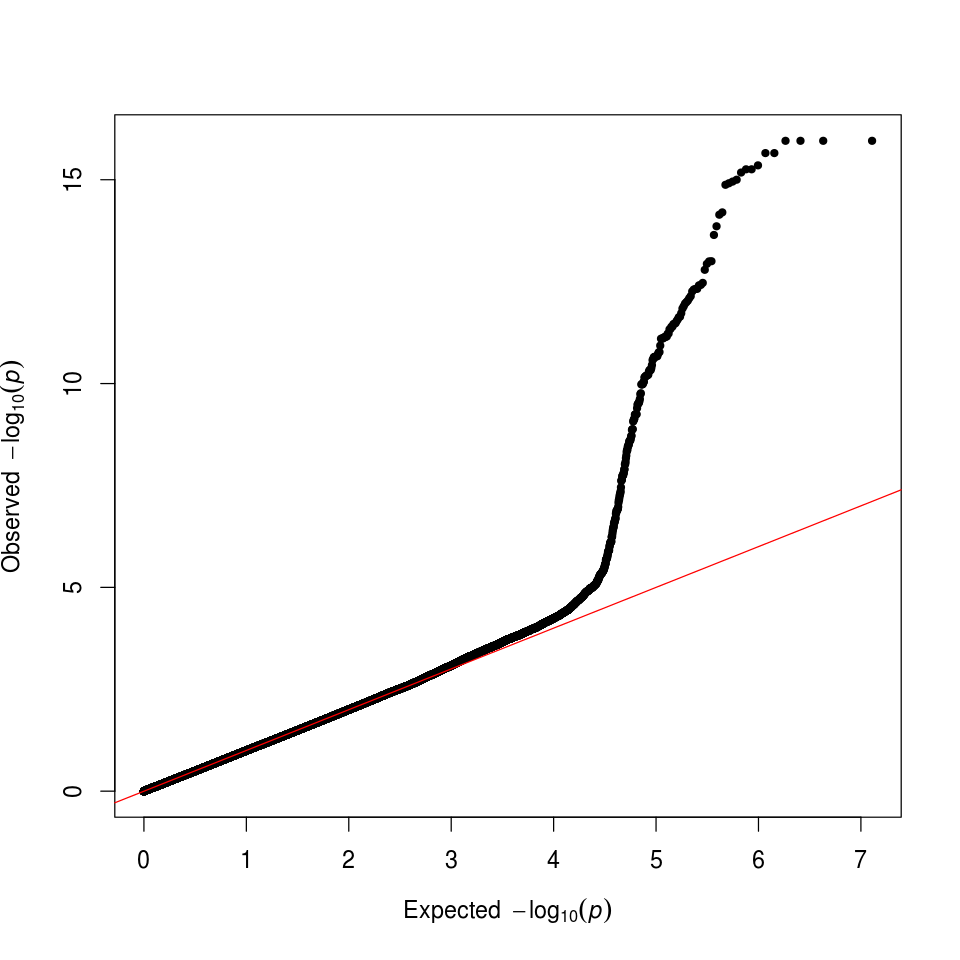
図8. GWAS Q-Qプロット
最後に、MLflowを使って、GWASの実行に用いたパラメーター、メトリクス、プロットを記録し、これによって解析の追跡、モニタリング、再現を可能にします。
解析のまとめ
このブログでは、Apache Spark、Delta LakeとMLflowを使ってGWASワークフローを端から端まで実行した結果をご覧いただきました。Sanford Health社の事例のような臨床における遺伝子型アッセイの精度検証やRegeneron社の事例のような標的遺伝子同定のためのGWAS解析結果のメタアナリシスでわかるように、Databricksのプラットフォームは、発展的な分析を容易にしたり、コードを書かない実験研究者や臨床家が大規模なデータセットを素早く探索するための、弊社製品付属のダッシュボード作成機能や、TableauやPowerBI といったBIツールへの最適化されたコネクターを通じて、下流解析における探索的な可視化を可能とします。
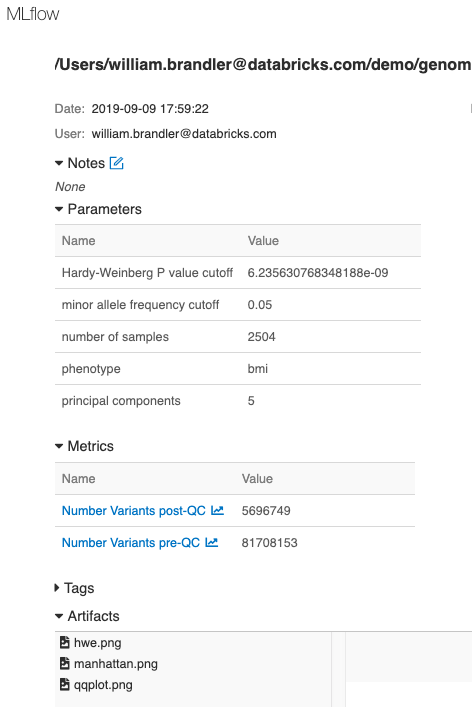
図9. MLflowによる各実行の追跡機能は、解析の再現性を確保します
GWASワークフローを端から端まで堅固に工学的に構築することで、研究者はフラットファイルのアドホックな分析から脱却して実環境での拡張性と再現性のある計算処理フレームワークに移行できます。さらに、VCFデータをDelta LakeにSparkデータフレームとして読み込むことで、研究者は表形式の表現型データ、EHR(電子カルテ)からのデータ、画像、リアルワールドエビデンスと実験計測値を統一されたフレームワーク上で統合することが可能になります。さあ今日から、工学的な集団レベルのGWAS Databricksノートブックをダウンロードして試してみましょう。
試してみよう!
Databricksプラットフォーム (Azure | AWS)上で、拡張性のあるGWASワークフローを実行してみましょう。