ヒト癌細胞データベース(CCLE)の解析
バイオデータセットの最低サンプル数
今回はアメリカの 「The Broad Institute」 が提供している、ヒト癌細胞の 「遺伝子発現データベース、CCLE」 の解析を行います。バイオビッグデータを解析していて困るのは、通常の機械学習と比べて 「データ数よりもパラメータ数の方が圧倒的に多いことが多い」 、ことです。
機械学習の大御所、 スタンフォード大学Andrew Ng先生の**「Machine learning」講義でも触れられていますが、基本的には 「It's not who has the best algorithm that wins,It's who has the most data」 が示す通り、データ数が少ない場合は、機械学習の精度に大きく貢献するのはアルゴリズム云々よりも 「データ数」** の方です。
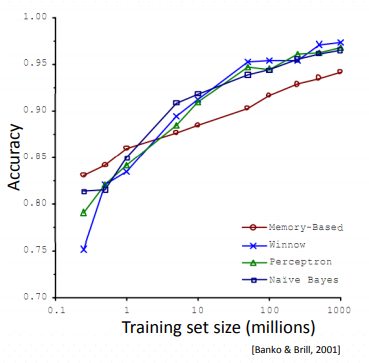
[Banko & Brill, 2001]
[Coursera:機械学習]
https://ja.coursera.org/learn/machine-learning
では 「どれくらいのデータ量が必要となるか」 、ということですが、上記の2001年の論文ではよく見るとX軸となるトレーニングデータセットのサイズの最小値が 0.1 millions(=100,000) となっています(!)。2001年当時はアルゴリズムが今ほど洗練されていなかったことを考慮しても、バイオデータセットで10万とか普通ムリ...これって機械学習的感覚からすると、よく囁かれる 「医学生物学論文の70%以上が再現できない」 のなんて当たり前じゃない?、だってデータセット(=モデル数)少なすぎるじゃん!、となる気がします。
私は元々バイオベースの人間なので、Andrew Ng先生のWeb講義受けて一番衝撃だったのが実はこのデータでした・・・この講義、個人的には全国の生物系大学の基礎学習課程で必須にした方がいいと思います。既存の堅固に見える生物学的ロジックが、数量的には砂上の楼閣に近いという感覚を初期に持つことは結構大事なんじゃないでしょうか・・・
(*最近は転移学習の研究も進んでいるため、Deep learningを用いて少数データ数の精度を上げよう、という流れもあると思うのですが、そのあたりはまだフォローしきれていません...)
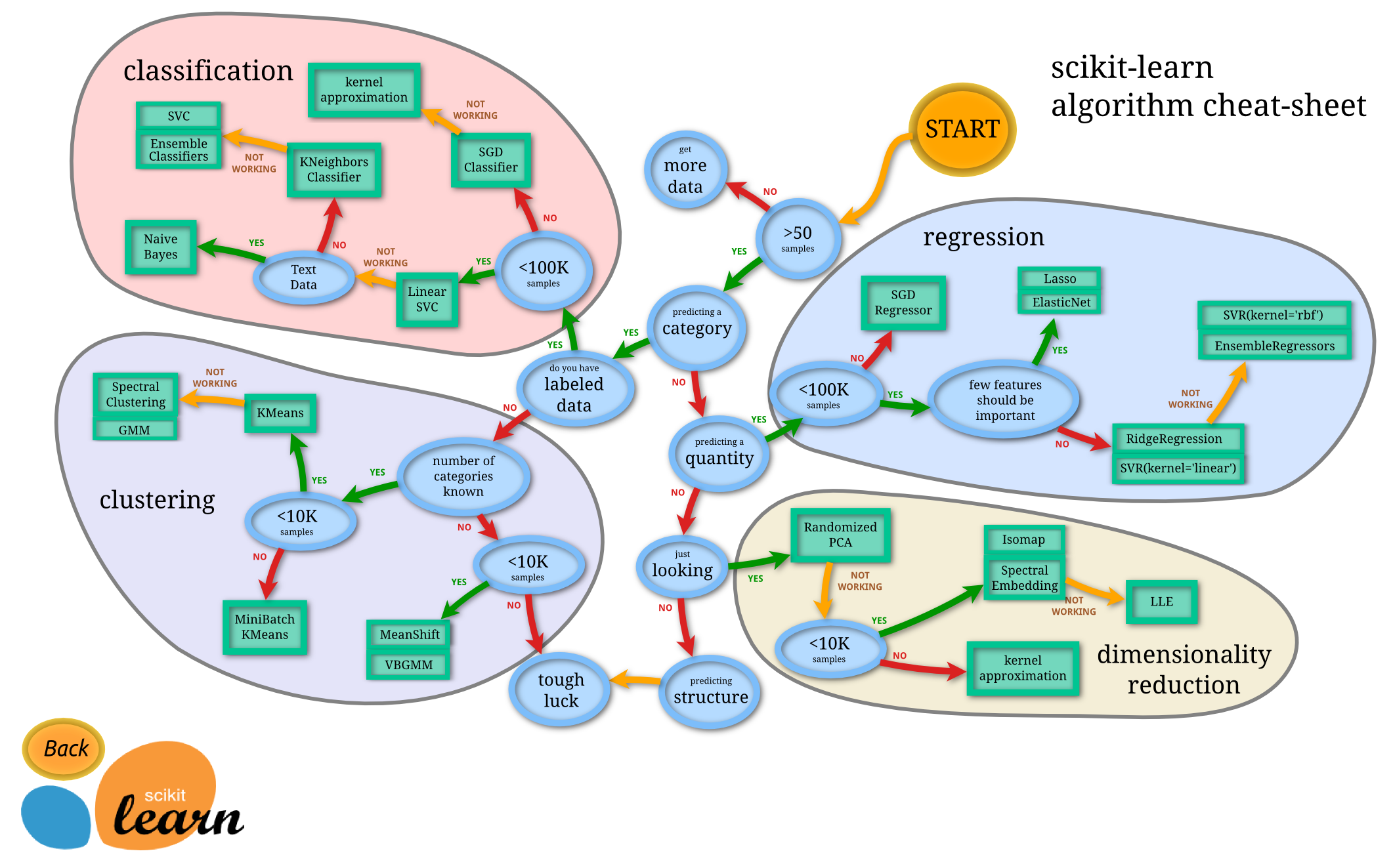
ただ、そうするとバイオインフォマティクスが全て終わってしまうので(笑)、気を撮り直してPythonの有名な機械学習ライブラリ、 「Scikit-learn」 のチートシートを見てみます。すると、一応ミニマムデータセットは 「50」 からに(ホッ)。
まあそれでも既存の生物研究のほとんどは(機械学習に適用するという意味では)消えてしまうのですが、今回の解析対象である 「CCLE」 は**「1019」**細胞なので、一応第一関門はクリアしたと個人的に納得して進めることにします(-0-)。
[Scikit-learn:機械学習チートシート]
https://scikit-learn.org/stable/tutorial/machine_learning_map/index.html
CCLEデータセットの取得
ヒト癌細胞の全遺伝子発現データセットを取得するにはCCLEのHPにアクセスし、上記左上の「Data」をクリックします。データ取得には登録が必要ですが、特に問題なく登録できます。
[CCLE:The Broad Institute]
https://portals.broadinstitute.org/ccle

そして「Data」ページの「Current Data」の項を見ると色々なデータセットがずらり・・・。
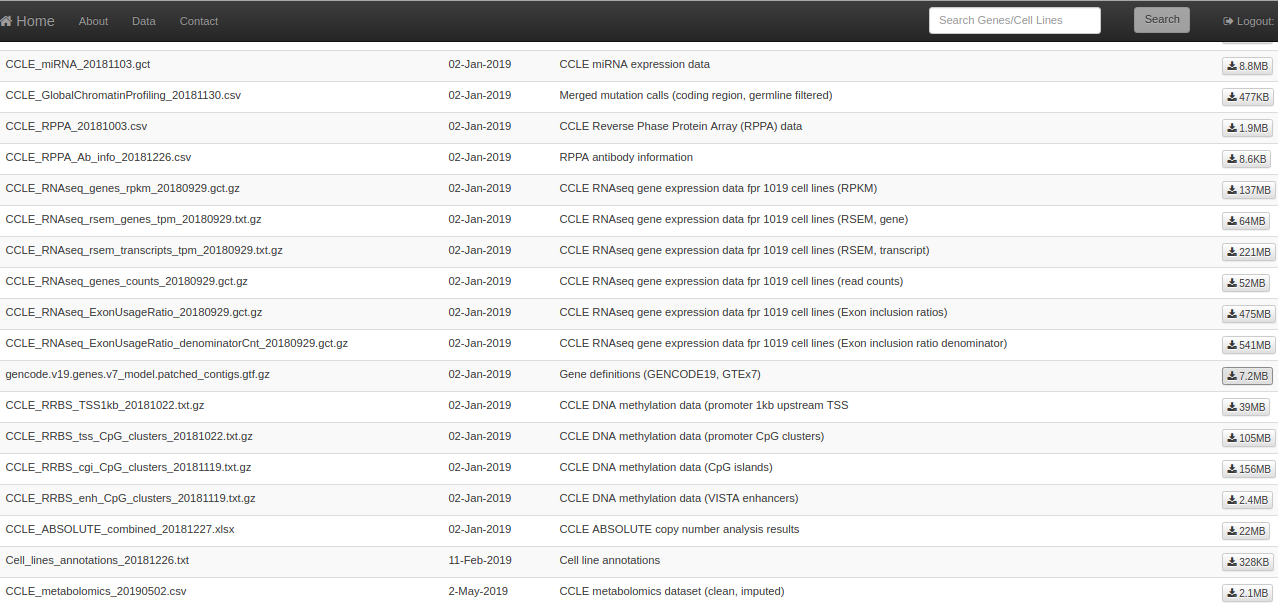
今回、遺伝子の発現情報の解析では、「RNA-seq」という手法で得られたデータを、「RSEM」というソフトウェアで解析し、「TPMカウント」にしたデータを使用します(ちなみにCCLEのLegacyデータには「Micro array」と手法で得られたデータも存在します。つまり、「遺伝子発現」と一口に言っても別種のデータが存在するんですね。これが実験科学の難しいところです)。
最近の知見では、TPMカウントは「Within-sample」(=似たような転写パターンを持つサンプル同士の比較)には良いが、「Between-sample」(=異なる転写パターンを持つサンプル同士の比較)には向いていないという話も聞きますが、私自身そこまで考慮して解析する自信がない・・・、ため、とりあえず「RPKM」よりは「TPM」の方が良いだろうという適当な感じで「TPM」データの解析を行うことにします。ちなみにこのあたりの込み入った話はチャンスがあればまた別の機会に・・・
[RNA-seq解析の手法]
https://bi.biopapyrus.jp/rnaseq/mapping/
https://bi.biopapyrus.jp/rnaseq/mapping/rsem/star-rsem-paired-rnaseq.html
https://bi.biopapyrus.jp/rnaseq/analysis/normalizaiton/tpm.html
CCLEから取得したデータは下記の(1)〜(3)種類です。
(1)CCLE_RNAseq_rsem_genes_tpm_20180929.txt.gz
解凍後> CLE_RNAseq_rsem_genes_tpm_20180929.txt
(2)Cell_lines_annotations_20181226.txt
(3)gencode.v19.genes.v7_model.patched_contigs.gtf.gz
解凍後> gencode.v19.genes.v7_model.patched_contigs.gtf
このうち、(2)はまあ特に取得する必要はないのですが、まあ一応念の為取得してみます。
(1)が遺伝子発現情報が入っているファイル、(2)がヒト癌細胞株のアノテーション情報、(3)が各遺伝子の情報が記載されているファイルです。
遺伝子ID情報の取得
このうち、まず(3)のファイルから必要な遺伝子のID情報を取得します。
awkを使っても良いのですが、なるべくPython純正でやりたいと思って 「GENCODE」 と呼ばれる、世界的にゲノム情報を集約しているグループのHPの一つを見てみると、 「gftparse」 と呼ばれるパッケージが使えるとの記述があったので今回はこれを使用してみます。
[GTF Parsers in Other Programming Language(GENCODE)]
https://www.gencodegenes.org/pages/data_format.html
https://github.com/openvax/gtfparse
https://anaconda.org/bioconda/gtfparse
[参考:awk を利用した GTF ファイルの処理]
https://bi.biopapyrus.jp/rnaseq/mapping/gtf.html
まずはターミナルでパッケージ「gtfparse」のインストール。
conda install -c bioconda gtfparse
その後の解析はjupyter notebookで行います。
Pandasとgtfparseのread_gtfをimport。
import pandas as pd
from gtfparse import read_gtf
次にダウンロード・解凍したgtfデータ(3)を読み込み、登録データの数・種類を見てみます。
df = read_gtf("gencode.v19.genes.v7_model.patched_contigs.gtf")
print(df.shape)
print(df.columns)
(436245, 21)
Index(['seqname', 'source', 'feature', 'start', 'end', 'score', 'strand',
'frame', 'gene_id', 'transcript_id', 'gene_type', 'gene_status',
'gene_name', 'transcript_type', 'transcript_status', 'transcript_name',
'level', 'havana_gene', 'exon_id', 'exon_number', 'tag'],
dtype='object')
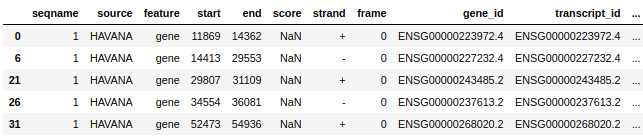
データ数は非常に膨大ですが、このうち今回の解析で必要なデータは遺伝子のIDのみなので、「feature」の種類で「gene」を絞り込んだあと、「gene_id」、「gene_name」、のデータのみに集約します。
```python
# featureの絞り込み
df_genes = df[df["feature"] == "gene"]
print(df_genes.head())
# 列の絞り込み
df2 = df_genes[["gene_id","gene_name"]]
print(df2.shape)
df2.head()
df_genes.head()
df2.head()
(56238, 2)

今回のデータセットでは遺伝子数が56238個のようですね。ちなみに、今回参照したCCLEのデータセットは少し古いゲノムをリファレンスとして使用しています。2019年5月現在、最新のヒトのリファレンスゲノムは「GRCh38」ですが、CCLEデータセットでは「GRCh37(UCSC version 19)」を使用しています。また、GENCODEとは別のグループに、リファレンスゲノム情報を集約している **「ENSEMBL」** がありますが、そこには過去にリリースされたリファレンスゲノムの一覧が記載されています。結構頻繁にリファレンスゲノムはupdateされているんですね。

例:2010年〜2014年ごろにかけたリファレンスゲノムのversionの移り変わり。

>[GENCODE: release history]
>https://www.gencodegenes.org/human/releases.html
>[ENSEMBL: Archives]
>https://www.ensembl.org/info/website/archives/assembly.html
話題が逸れましたが、とりあえず上記で集約したデータをcsvファイルにしておきます。
とりあえず長くなったので今回はここまで。
(たぶん近いうちに掲載予定)次回は、今回の集約データを用いていよいよ遺伝子情報の解析を実施していく予定です。
>```python
df2.to_csv("r_genome_20190526.csv")