はじめに
Rの生命科学情報のパッケージを集めたBioconductorにenrichplotというライブラリがあります。
enrichplotを使うことでエンリッチメント解析の結果をグラフに表示することができます。
作者は、中国広州市の南方医科大学の余光创(Guangchuang Yu)さんで、近年clusterProfilerなどの有名なRのパッケージを多数作成されています。
上のチュートリアルで説明されているように、cnetplot関数を呼ぶことで、下記のようなパスウェイと遺伝子を含むグラフを作成することができますが、
library(enrichplot)
library(DOSE)
data(geneList)
de <- names(geneList)[abs(geneList) > 2]
edo <- enrichDGN(de)
edox <- setReadable(edo, 'org.Hs.eg.db', 'ENTREZID')
p1 <- cnetplot(edox, foldChange=geneList)
## categorySize can be scaled by 'pvalue' or 'geneNum'
p2 <- cnetplot(edox, categorySize="pvalue", foldChange=geneList)
p3 <- cnetplot(edox, foldChange=geneList, circular = TRUE, colorEdge = TRUE)
cowplot::plot_grid(p1, p2, p3, ncol=3, labels=LETTERS[1:3], rel_widths=c(.8, .8, 1.2))

図がビジーになってしまいがちなので、自分の興味があるパスウェイに絞り込みたい場合があります。
showCategory は文字列ベクトルも引数にとれる
ソースコードを見ると、ドキュメント化されていない showCategory 引数があり、これらは通常、5 などのアトミックベクトルを引数に取りますが、文字列ベクトルなども引数に取ることができることがわかります。(utility.Rのextract_geneSets, update_n 関数参照)
そこで、showCategory = c("Basal-Like Breast Carcinoma","Inflammatory Breast Carcinoma") などとパスウェイのDescriptionを指定して
library(enrichplot)
library(DOSE)
data(geneList)
de <- names(geneList)[abs(geneList) > 2]
edo <- enrichDGN(de)
edox <- setReadable(edo, 'org.Hs.eg.db', 'ENTREZID')
p1 <- cnetplot(edox, showCategory = c("Basal-Like Breast Carcinoma","Inflammatory Breast Carcinoma"),
foldChange=geneList)
## categorySize can be scaled by 'pvalue' or 'geneNum'
p2 <- cnetplot(edox, showCategory = c("Basal-Like Breast Carcinoma","Inflammatory Breast Carcinoma"),
categorySize="pvalue", foldChange=geneList)
p3 <- cnetplot(edox, showCategory = c("Basal-Like Breast Carcinoma","Inflammatory Breast Carcinoma"),
foldChange=geneList, circular = TRUE, colorEdge = TRUE)
cowplot::plot_grid(p1, p2, p3, ncol=3, labels=LETTERS[1:3], rel_widths=c(.8, .8, 1.2))
とすれば、下図のように表示するパスウェイを絞り込むことができます。
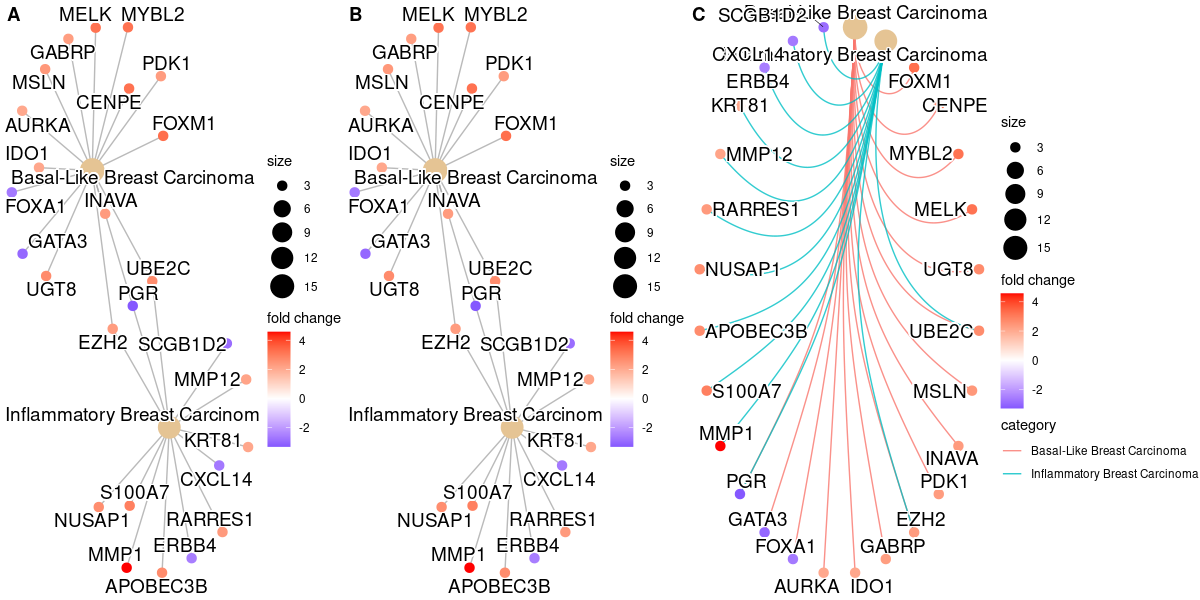
指定された2つのパスウェイのみが表示されました。
この記事は以上です。