Qiita はオミクスデータを解析・管理できるプラットフォーム
前回はQiita の説明と利用登録,サンプル情報の追加を行いました.
今回は実際に16Sアンプリコン配列データをアップロードし下処理していきます.
生データ (fastqファイル)を追加する
サンプル情報とデータを関連付けさせるためにprep information という用いたプライマーやシーケンサの種類を記載した技術的な情報を追加しなければいけません.
prep information についてはQiita のドキュメントに詳しく書かれています.
「Upload Files」にてprep_information_16S.txtをアップロードします.
そして,「Add New Preparation」にて「Select file:」でprep_information_16S.txtを選択,「Select data type:」で「16S」を選択します.
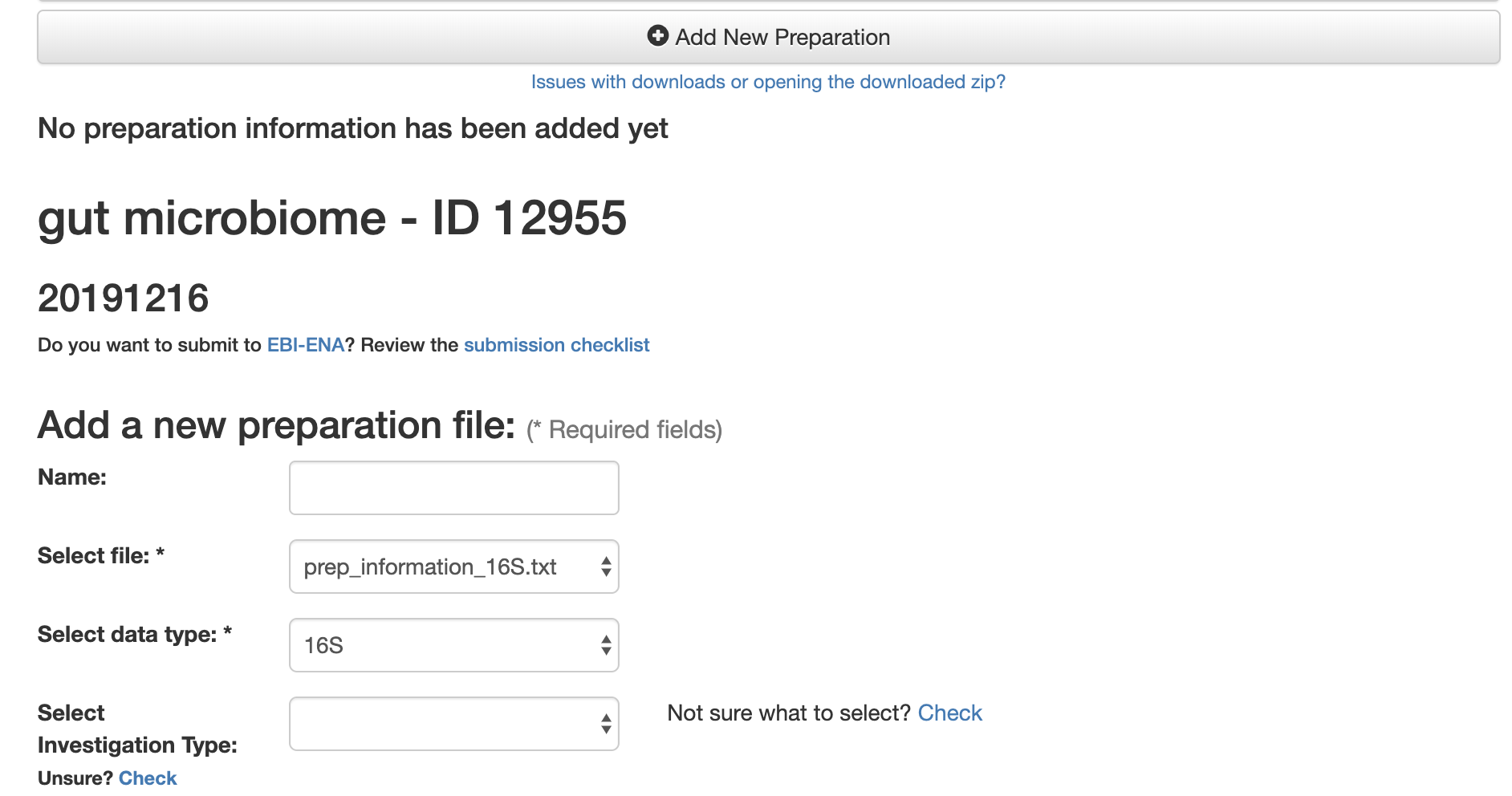
またこのタイミングで16S/に入っているfastq.gz ファイルも全部「Upload Files」にてアップロードしておきます.
「Data Types」に「16S」が追加されるのでそこをクリックします.
そして,「Select type:」で「per_sample_FASTQ」を選択します.
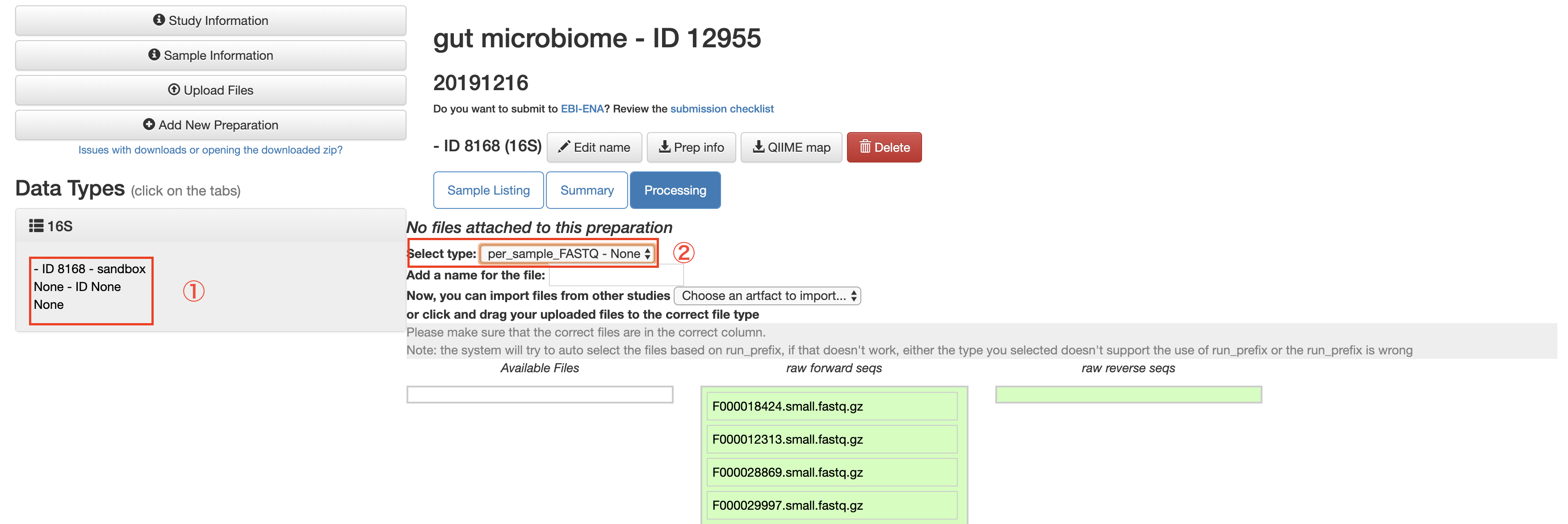
「Add a name ofr the file:」に任意の名前を入力し「Add files」をクリックすることで,このfastqファイルのセットに名前をつけます.
そうすると以下のように,ファイル処理プロセスのネットワークが表示されるようになります.
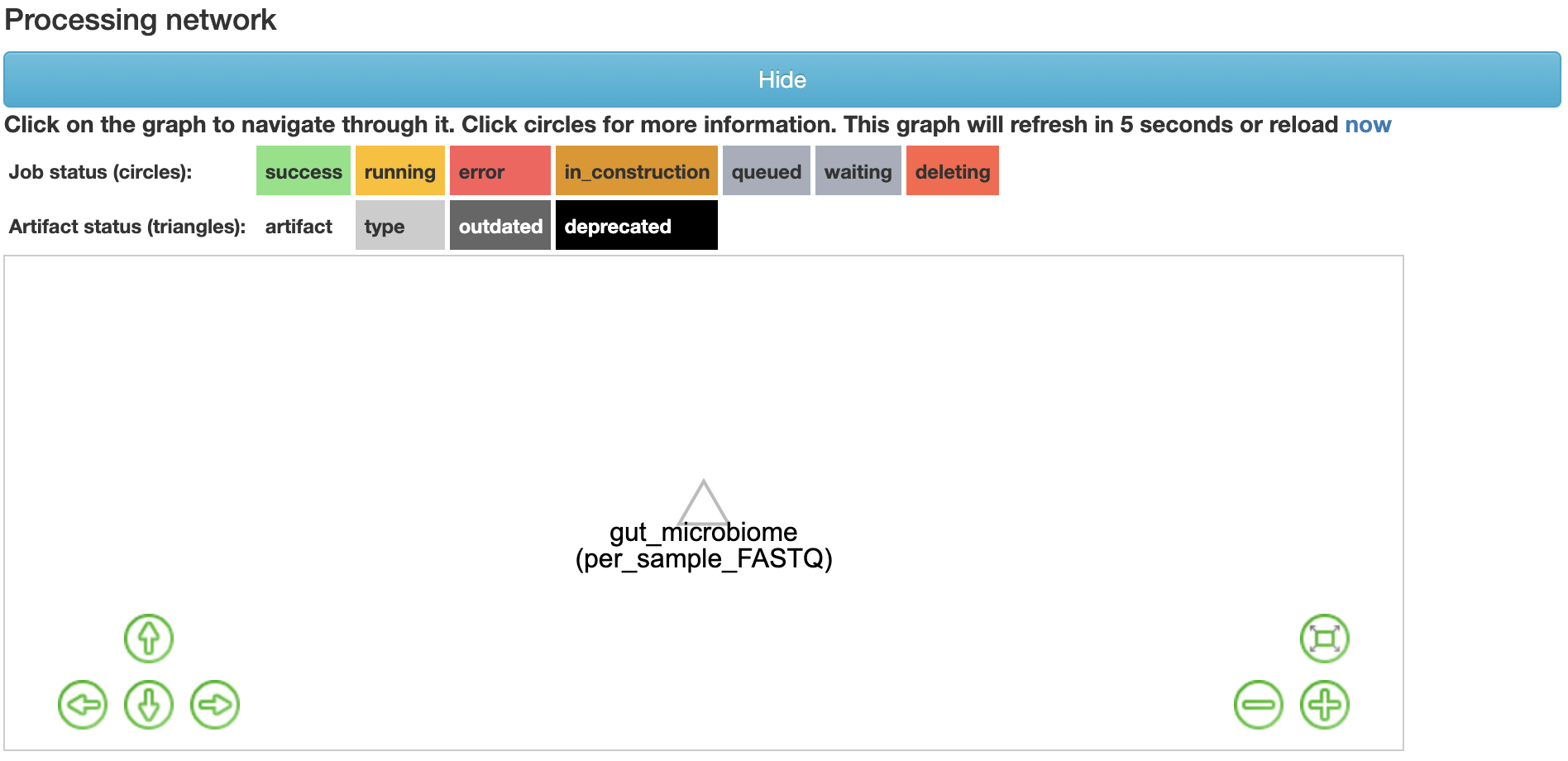
データの処理
ライブラリの分割
「per_sample_FASTQ」オブジェクトをクリックします (上の画像の△のやつです).
そうすると,「Choose command:」が出てくるので,以下のように「Split libraries FASTQ」を選択します.
「Parameter set:」にて「Per-sample FASTQs; phred_offset 33」を選択すると,詳細なパラメータ選択が出てきます.

パラメータを確認したら「Add Command」をクリックしましょう.
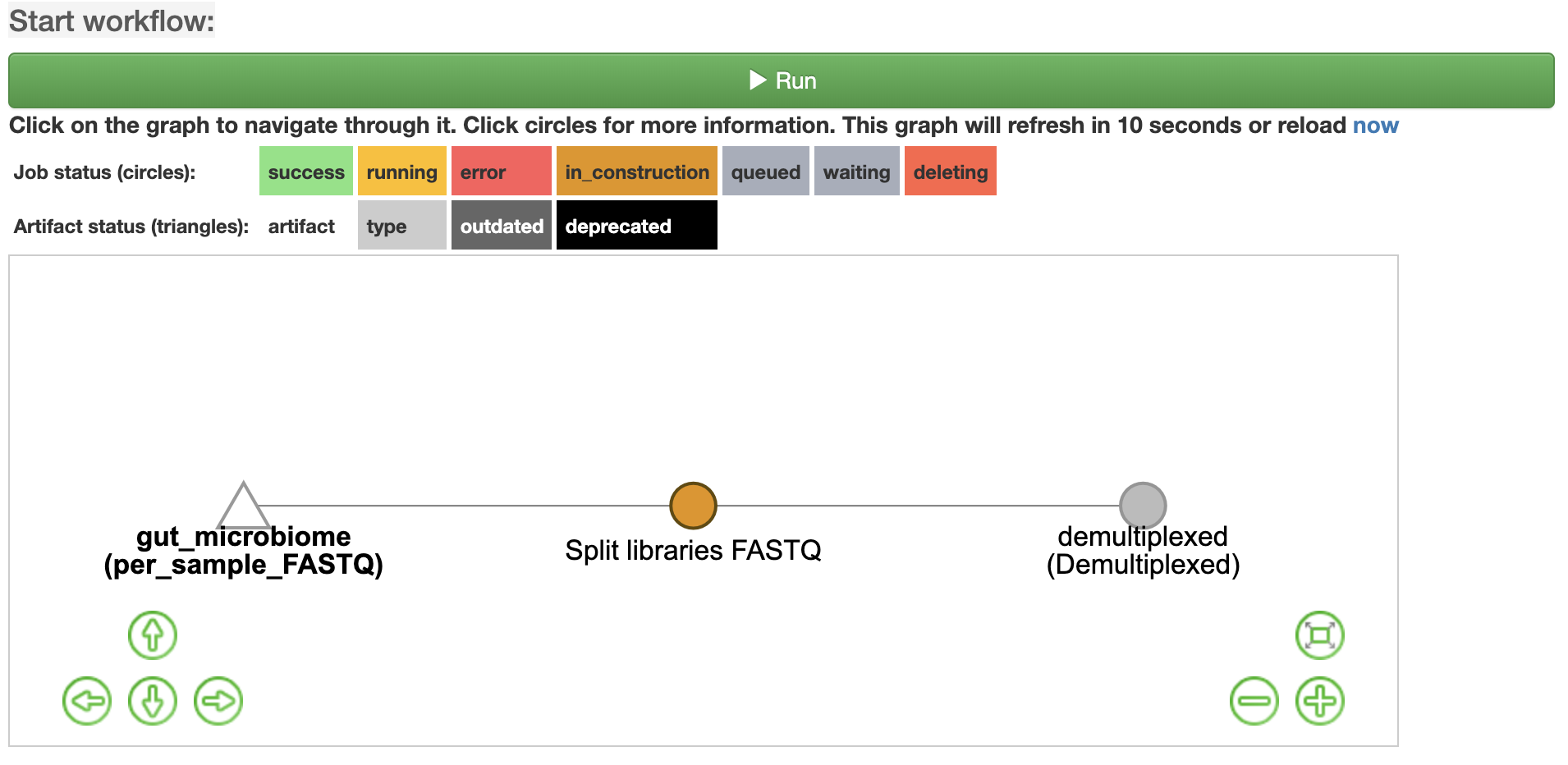
そうすると,このようにファイルネットワークが更新されます.
「Start workflow:」で「Run」を押せば実行されますが,まだ押しません.
他の下処理プロセスも追加して,最後にまとめて「Run」しましょう.
deblur によるエラー修正
delur はqiime2 などでも使われているシーケンス配列のエラー修正をしてくれるツールで,クオリティスコアに基づいてフィルタリングもしてくれます.
今回は,Single-end のfastqファイルを処理するのでdeblur ですが,Paired-end のfastqファイルを処理するときには,DADA2などを使います.
先に「Choose command:」で「Trimming」を選択し配列を同じ長さにするとdeblur が機能しやすくなるようです.
Although you can deblur the demultiplexed sequences directly, deblur works best when all the sequences are the same length.
Qiita-GNPS-workshop The deblur workflow
今回はV4配列を使っていますので,100bpでトリミングしてみます.
パラメータを確認し「Add Command」をクリックです.
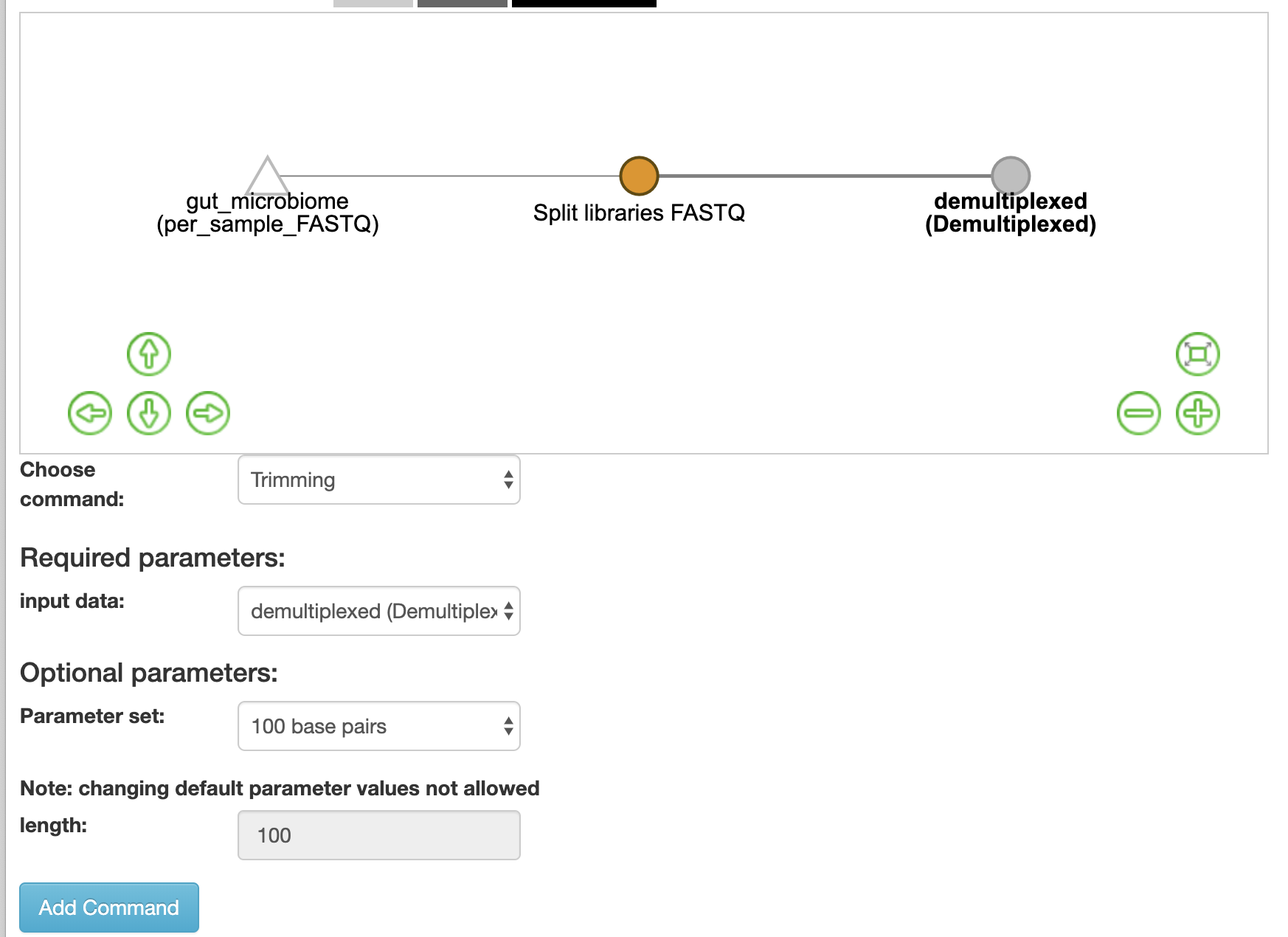
またファイルネットワークが更新されました.
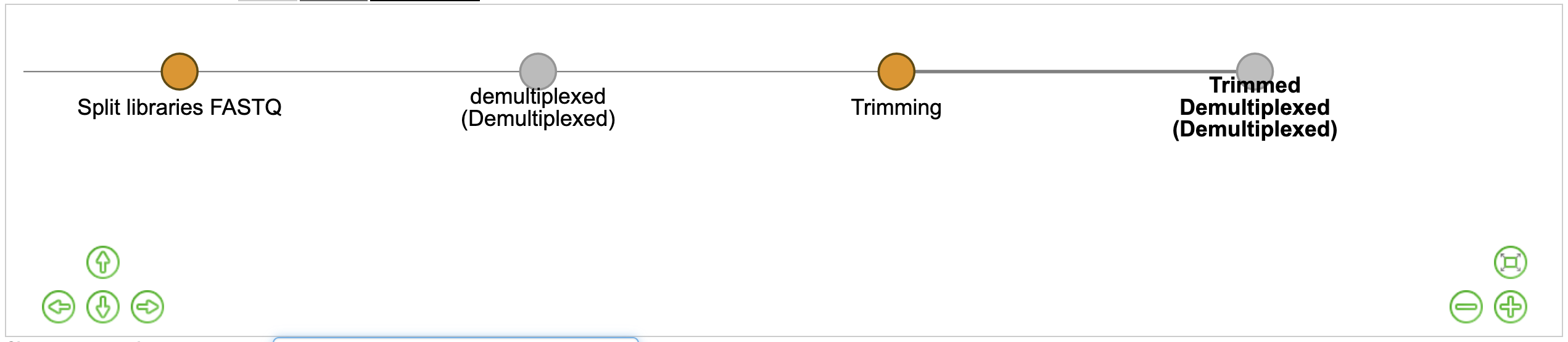
次に,「Trimmed Demultiplexed」をクリックし,「Choose command:」にて「Deblur」を選択します.
細かいオプションを決めてまた「Add Command」です.

またファイルネットワークが更新されました.
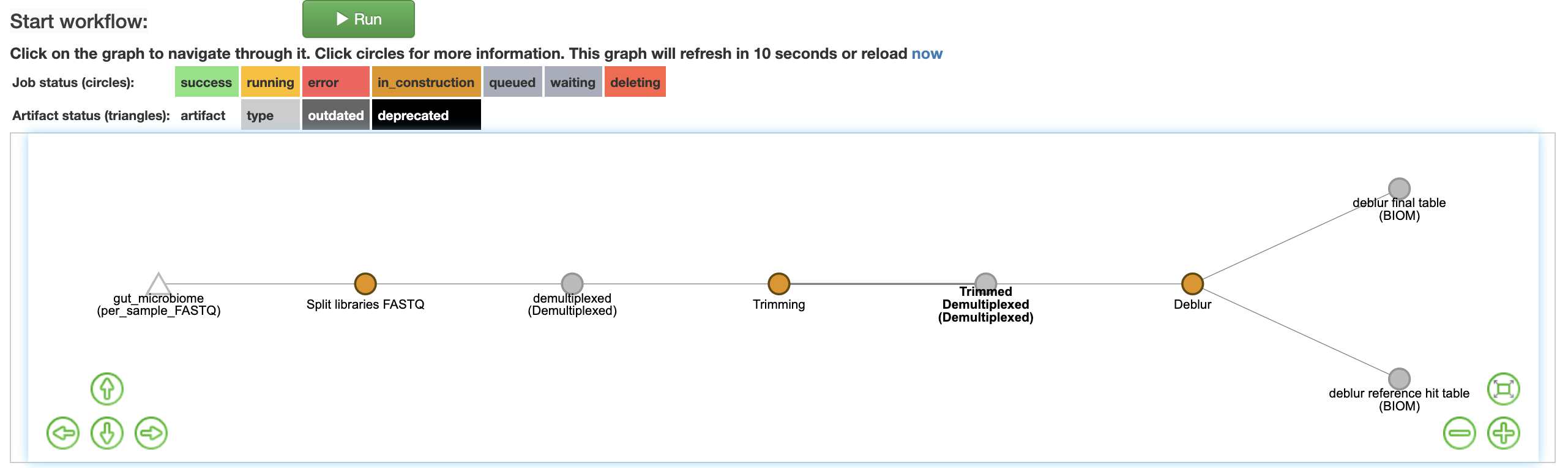
最終的に作ったファイルネットワークは以下のようになりました.
「Run」をクリックし,作ったワークフローを動かします.
おさらい
今回は,Qiita にfastqファイルを追加し,各種下処理コマンドを追加していきファイルネットワークを作りました.
ファイルネットワークでオレンジの丸で示されている通り行う処理は
- Split libraries FASTQ
- Trimming
- Deblur
という3工程になっています.
次回は,実際に下処理が終わったファイルを解析していきます.