GRiDとは
**Growth Rate InDex (GRiD)**のことで,本指数はメタゲノム配列のin situ 微生物増殖速度を推定するために Emiolaらによって2018年に開発されました (論文).
そもそもなんでメタゲノム配列から微生物の増殖速度を知ろうとしているかというと論文いわく,
「ショットガンシーケンスは複雑な微生物の組成と機能を調べる強力なツールだよね」
↓
「だけどこの手法,異なる環境条件下や疾患間において微生物の増殖速度が変化する可能性のある微生物群集の複雑な動的性質を反映できないよね」
↓
「各微生物の増殖速度が推定できれば,ある環境下の微生物群集の表現型に大きく寄与している微生物たちの正体がわかるよね」
↓
「さらに,急速に増殖する細胞と静止している細胞の比率を推定すれば,拮抗する微生物種間の相互作用もわかるよね」
ということのようです.
メタゲノムから各細菌の増殖速度を推定するにはiRep というツールがありますが,メタゲノム配列高いカバレッジ (>5x)が要求されます.皮膚などヒトの微生物群集のゲノムでは平均して5%未満しかその基準に達しないようです.
GRiD は超低カバレッジ (0.2×, which is roughly equivalent to 0.05% relative abundance of a 2.5 Mbp genome from a metagenomic sample of 100 bp x 10 million reads) なメタゲノム配列から微生物増殖速度を推定できます.
また,論文では「de novo アセンブルしたメタゲノム配列の増殖速度推定」と言っていますが,ツールではfastq 形式のファイルも解析することができ,アセンブルに依存しない解析もできます.
本記事では,公式Github の記事を参考に作成しています.
インストール
GRiD はいくつかのツールと依存関係があるので,miniconda を使用するとよいです.
# Set up channels
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
# Install GRiD
conda install grid=1.3
# As an example, you can create an environment for GRiD.
conda create --name GRiD
source activate GRiD
conda install grid=1.3
コマンドの紹介
主に使うコマンドは以下のとおりです.
grid single <options>
grid multiplex <options>
grid -v
grid -h
grid single : 単体の微生物ゲノム配列を用いるとき
grid multiplex : メタゲノム配列を用いるとき (別途GRiDデータベースが必要)
grid -v : バージョンの確認 (2019.12現在は1.3が最新のようです)
grid -h : オプションなどのメッセージ確認
GRiD ではfastqファイルも受け付けていますが,Paired-end ファイルは1つのファイルに結合する必要があります.
テストデータを試してみる
テストデータには_Staphylococcus epidermids_, Lactobacillus gasseri, _Campylobacter upsaliensis_のゲノム配列がカバレッジ0.5程度ずつ含まれています.
# Download the data
wget https://github.com/ohlab/GRiD/archive/1.3.tar.gz
cd GRiD-1.3/test
# Run single module
# Staphylococcus epidermidis
grid single -r . -g S_epidermidis.LRKNS118.fna -o output_single
# Run multiplex module
grid multiplex -r . -d . -p -c 0.2 -o output_multiplex -n 16
解析が終わると,PDFとテキストが出力されます.
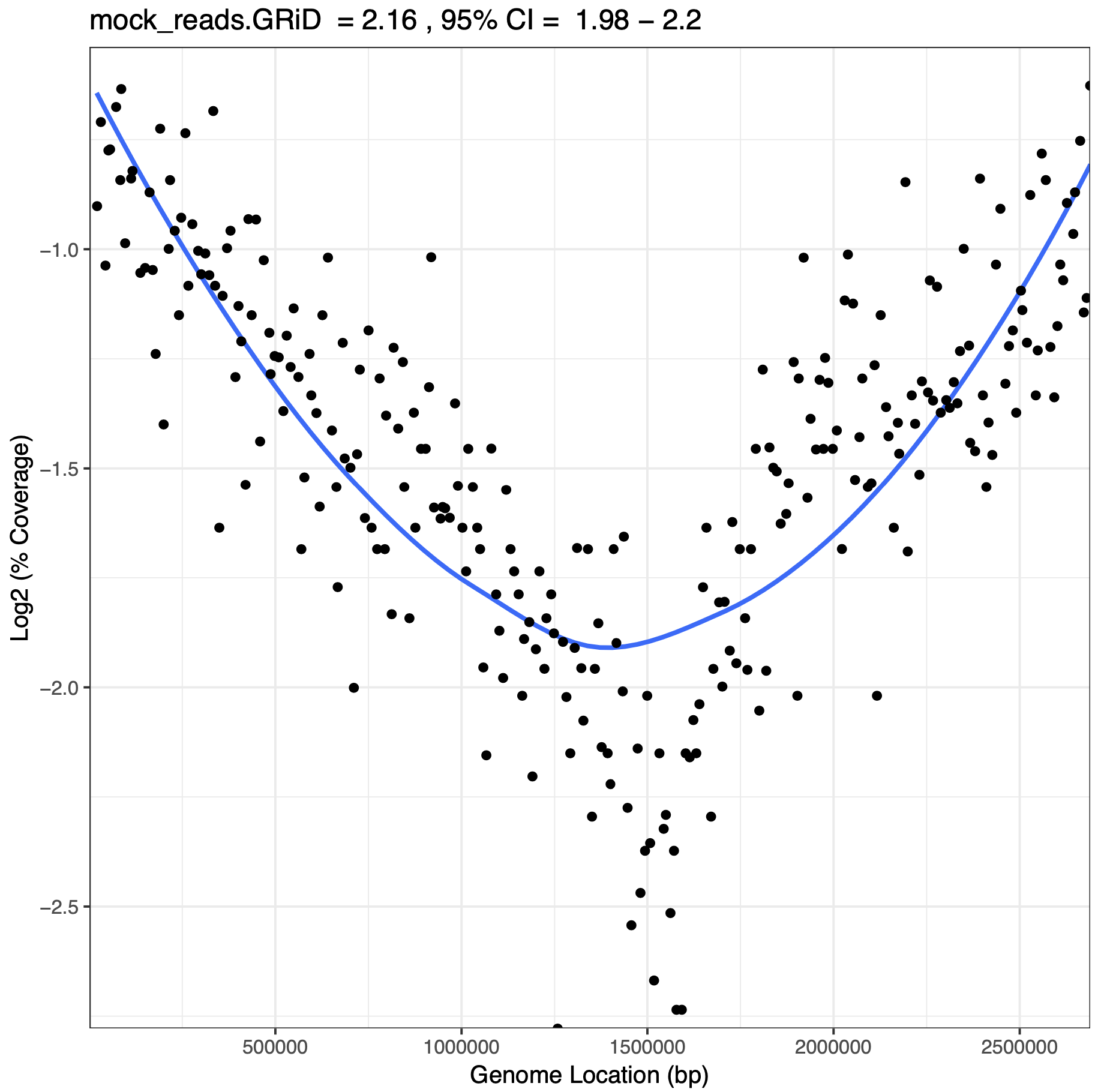
細菌ゲノム配列 (Staphylococcus epidermidis)に対する解析結果
ほとんどの細菌ゲノムにおいて_dnaA_ はori 付近にある一方で,ゲノムの複製終結点は一般的に_dif_ 配列付近になります.つまり,dnaA/ori ratio とter/dif ratio が1に近似するほどGRiDスコアの精度が高いと言えます.
| Sample | GRiD | 95% CI | GRiD unrefined | Species heterogeneity | Coverage | dnaA/ori ratio | ter/dif ratio |
|---|---|---|---|---|---|---|---|
| mock_reads.GRiD | 2.16 | 1.98 - 2.2 | 2.41 | 0.103734439834025 | 0.557 | 0.905614270624413 | 0.936734611636753 |
こちらはゲノムの位置とカバレッジ情報を示す散布図です.
メタゲノム配列に対する解析結果
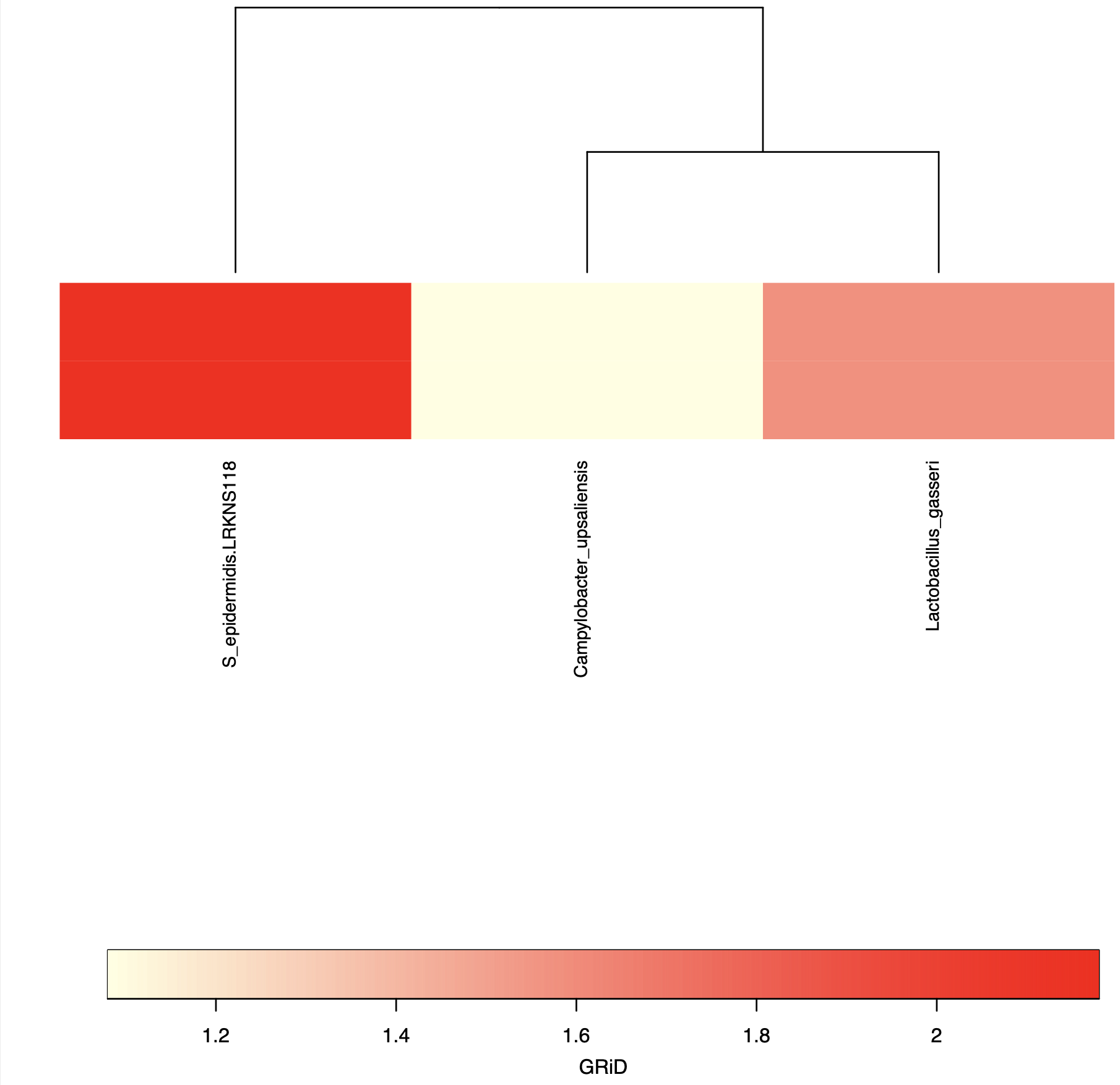
メタゲノム配列では,配列に含まれる細菌ごとにGRiD スコアやカバレッジ情報が出力されます.
| Genome | GRiD | GRiD_unrefined | Species_heterogeneity | Coverage |
|---|---|---|---|---|
| Campylobacter_upsaliensis | 1.08 | 1.39 | 0.223021582733813 | 0.477 |
| Lactobacillus_gasseri | 1.58 | 1.81 | 0.12707182320442 | 0.566 |
| S_epidermidis.LRKNS118 | 2.18 | 2.44 | 0.10655737704918 | 0.556 |
ヒートマップと階層的クラスタリングの組み合わされた図が出ます.
今回の場合,3株のうち_S_epidermidis.LRKNS118_ が最も増殖速度が速いようです.
実際に手元にあるメタゲノムデータ (10GB*20ファイル程度)で試したところ2ヶ月程度かかりました.
CPU48コア,RAM500GB程度のマシンでしたが内部でRを動かしているからかかなり時間がかかりました.
大きいメタゲノム配列を解析したい場合,いくつかの方法でgrid multiplex モジュールのパフォーマンスを向上できます.
-
environ_specific_databaseというGRiDデータベースを用いるか,独自で作成したデータ量の少ないカスタムデータベースを用いる.
FTPサイト から糞便用,皮膚用のデータベースをダウンロードできます. -
メタゲノム配列をサブサンプリングして,解析するデータ量を削減する.
-
Species heterogeneityに基づいて結果をフィルタリングする.
公式Github によると0.3 未満になると信頼性が非常に高くなるようです.
データベースの作成と更新
解析にはGRiDデータベースが必要なので作成しておきます.
また,一定期間が過ぎたものは更新します.
update_database <options>
<options>
-d GRiD database directory (必須)
-g Bacterial genomes directory (必須)
-p Prefix for new database (必須)
-l Path to file listing specific genomes
for inclusion in database [default = include all genomes in directory]
-h Display this message
※ゲノムはfasta形式である必要があります
依存関係
もしGRiD を野良ビルドした方は注意してください.
公式が実行テストしたものには,"tested" と記載があります.
-
R (tested using v 3.4.1)
- Required R libraries - dplyr, getopt, ggplot2, gsubfn, gplots
-
bowtie2 (tested using v 2.3.1)
-
seqtk (tested using v 1.0-r31)
-
samtools (tested using v 1.5)
-
bedtools (tested using v 2.26.0)
-
bamtools (tested using v 1.0.2)
-
blast (tested using v 2.6.0)
-
Pathoscope2
-
parallel
-
mosdepth