Roary
今回はRoaryでバクテリアの比較ゲノム解析をしてみます。Part.1, Part.2の続きになります。
Roary公式サイト: https://sanger-pathogens.github.io/Roary/
Roaryって何?
公式サイトによれば、
By Andrew Page based on version 3.11.2 (22-Jan-2018)
Roary is a high speed stand alone pan genome pipeline, which takes annotated assemblies in GFF3 format (produced by Prokka (Seemann, 2014)) and calculates the pan genome. Using a standard desktop PC, it can analyse datasets with thousands of samples, something which is computationally infeasible with existing methods, without compromising the quality of the results. 128 samples can be analysed in under 1 hour using 1 GB of RAM and a single processor. To perform this analysis using existing methods would take weeks and hundreds of GB of RAM. Roary is not intended for meta-genomics or for comparing extremely diverse sets of genomes.
となっており、要はそんなに高いスペックがなくても、高速でパンゲノム解析できるツールである。通常、パンゲノム解析には非常に高度なコマンド操作技術と高級なPCが必要であった。それを極めて簡単にし、誰でもできるようにしたのがRoaryである。他にもパンゲノム解析ソフトはあるが、当方としてはRoaryが最も使いやすい印象を受けている。
使い方
前回までにやったことはProkkaでゲノムに遺伝子情報をアノテーションしたところまでであるので、続いてProkkaが作成したフォルダの中にあるGFF3ファイルを取り出し、これを使ってゲノム比較をRoaryで行う。GFFファイルはTranscriptを含めた遺伝子情報が記載されたファイルと考えてほしい。詳しくは上坂先生のサイトを参照。
まずは、Prokkaで出力されるファイルは日付を元に名前が決められるようなので、サンプル名依存にしたい。(例: PROKKA_12242019.fna) そこで、forとmvコマンドを使って変えていく。ここで決めた名前に応じて、系統樹の時の名前が決められる。今回はGCFから始まる番号で名前をつけておく。
Roaryを日本語で紹介しているサイトはあまりないので、今回は-hで出てくるヘルプも載せておく。Roaryにもたくさんのオプションがある。
Usage: roary [options] *.gff
Options: -p INT number of threads [1]
-o STR clusters output filename [clustered_proteins]
-f STR output directory [.]
-e create a multiFASTA alignment of core genes using PRANK
-n fast core gene alignment with MAFFT, use with -e
-i minimum percentage identity for blastp [95]
-cd FLOAT percentage of isolates a gene must be in to be core [99]
-qc generate QC report with Kraken
-k STR path to Kraken database for QC, use with -qc
-a check dependancies and print versions
-b STR blastp executable [blastp]
-c STR mcl executable [mcl]
-d STR mcxdeblast executable [mcxdeblast]
-g INT maximum number of clusters [50000]
-m STR makeblastdb executable [makeblastdb]
-r create R plots, requires R and ggplot2
-s dont split paralogs
-t INT translation table [11]
-ap allow paralogs in core alignment
-z dont delete intermediate files
-v verbose output to STDOUT
-w print version and exit
-y add gene inference information to spreadsheet, doesnt work with -e
-iv STR Change the MCL inflation value [1.5]
-h this help message
ベースとなる使い方は以下のようである。UNIXにおいて*は何でもという意味。GFFファイルがあるディレクトリをカレントディレクトリにして、以下のコマンドを打つと全てのファイルを計算に含んで比較してくれる。
roary -e --mafft -p 8 *.gff
この方法で出力されるファイルを利用するとアミノ酸レベルでの違いを見ることができたりするが、普通SNPを確認したりしたくなるものなので、その場合のやり方を紹介する。と言っても、Roaryは本来計算過程でSNPを確認している。なので、Intermediateとして生じるファイルを消さないようにしておけば良い。したがってコマンドとしては、
roary -e --mafft -z *.gff
とかになる。-mafftを指定しないと計算がゆっくりになる。どちらが正確とかの議論は詳しくないので置いておく。-zで全ての計算過程のファイルが残されるので、その際に生じるmultiFASTAファイルも残すことができ、それをSnapGeneなどで確認するとSNPが確認できる。解析時間は数時間といったところで、やはり非常に計算は早い。手元の適当なデータの解析結果になるが、SnapGeneで出力されたファイルを確認すると、下図のようになる。おそらく共通する遺伝子に対して全てSNP検索をかけてくれるので、もしも近縁な種でSNPを知りたい場合は有効活用できそうである。Intermediateファイルはpan_genome_sequencesフォルダにある。注意点としては関係ない種を入れるとコアゲノムの数が減る点であろうか。
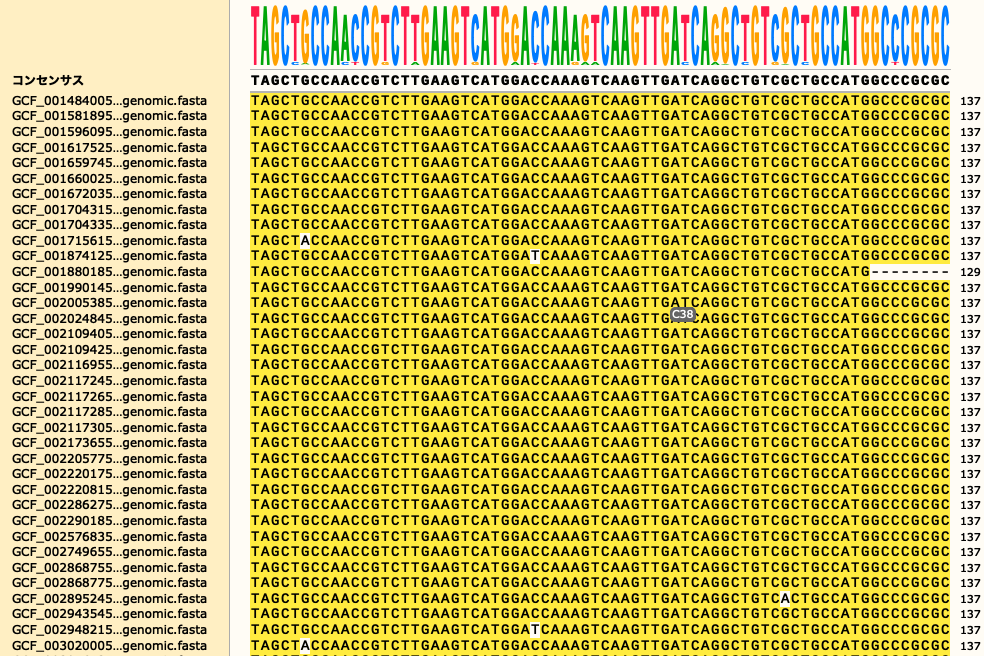
一応、出力されるファイルとしては summary_statistics.txt, gene_presence_absence.csv, gene_presence_absence.Rtab, pan_genome_reference.fa, accessory_binary_genes.fa.newick, accessory_graph.dot, core_accessory_graph.dot, clustered_proteins, core_gene_alignment.alnなどがある。https://sanger-pathogens.github.io/Roary/ を参考にPythonのスクリプトを作っておくと、さらに3枚のフィギュアが作れる。Rtabファイルはそのまま見れないので、R上でhttps://github.com/sanger-pathogens/Roary/blob/master/bin/create_pan_genome_plots.R からRスクリプトを作成して、作図する。よく確認すべきファイルはgene_presence_absence.csvになる。遺伝子の有る無しをインプットした株ごとに確認できる。比較ゲノムで重要な点が詰まっていそうである。newickファイルはMEGA Xで開くと系統樹になる。
まとめ
Roaryはすごい。早い。優しい。
Roaryシリーズ
Part.1: https://qiita.com/danryo_official/items/fe9273f13368ac5ad780
Part.2: https://qiita.com/danryo_official/items/3e9dc463daf9e35870e7
Part.3: https://qiita.com/danryo_official/items/11353fdf2700c9b6ffe3