初日から、超ネガティブなことを書いて大変申し訳無いが、この記事ではバイオインフォ分野で利用されている現状のワークフロー言語の足りないと思うところを紹介しようと思う。ワークフロー関係者からしたら、もしかしたらあまりいい気分がしないかもしれないが、立場が違えばそういう見方もあるということで参考として読んでもらいたい。
ワークフローに対する私見
なぜ人はワークフローを書くのかと言えば、それはデータ解析の再現性のためだろう。つまり、誰かがある環境で作ったツールや解析の実行手順を、他の環境でも再現できることを目的としており、そのために解析ツールの実行手順を決まった形式(ワークフロー言語)に従って書いている。またおそらく、その他の環境では、"自分以外の人間"(=他人)が、別のデータを使って同じ処理を行うことを想定している。ここで言う他人は、半年後の自分なども含んでいる。
そういったことを実現させるものとしては、各言語のパッケージも自由度の高いワークフローみたいなものだし(実際ワークフローパッケージというものもある)、Jupyter Notebookも別のデータを入力として再実行するやり方があるので(Papermill)、使いかた次第ではワークフロー的であるが、ここではバイオインフォ分野で利用されているMake、Rake、Snakemake、Nextflow、Swift、CWL...のようなワークフロー言語で、既存のツールが組み合わさった状態で公開されたワークフローを想定している。もう少し簡素なやり方としては、Docker、Singularityで、個々のツールごとにコンテナ化するというやり方もあると思う。あと、GUI上で解析ツールを繋げていくようなGalaxy、KNIME、BaseSpace、Terraみたいなアプリケーションもできあがったものはワークフローだし、また、こういう話しはちょっと昔はパイプラインという言い方がされていた気がする(PipelinePilot、Taberunaなど)。あと、JenkinsやTravisのような継続的インテグレーション(CI)系のツールも、テストというタスクに関してのワークフローでもある。
昨今バイオ業界での機運が高まるなか、個人的にはワークフローの話は聞いてて楽しくない(ワークフローで幸せになる人がいる事は認めるが)。これはそもそも、自分がツールを作る側の人間であるため、他人が作ったツールを公開しても、研究にはならなかったり、毎日新しいツールが出てくるスーパーレッドオーシャンな業界にいて、このツールを使っていれば良いみたいな状態にならない、立場的な要因が大きいのかもしれないが、ワークフロー関係者とはいつも何か温度差を感じていたので、その原因がどこにあるのかを考察してみた。
一般的なワークフローのイメージ
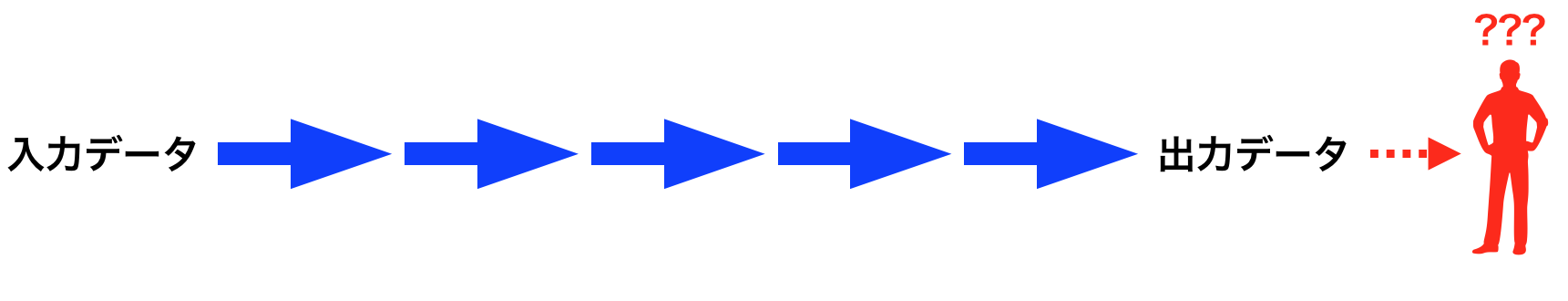
ワークフローの発表スライドではよくこういう矢印が連なったような図が出てくる。

あたかも、水の流れのように全てのステップがスムーズに実行され、ある入力に対して、テンポよく出力が得られるようなものを連想する。また途中ちょっとした分岐やマージくらいは想定しているかもしれない。しかし、自分は以下で示すように、そのようなワークフローをほとんど作ったことが無いし、他人のワークフローを実際のデータ解析で使ったこともない。
ケース1: データの再現性
まず言いたいことは、「ワークフロー化すれば、あらゆる環境でツールが動く」と言うのはワークフロー開発者側の方便だと思う。何故ならば、ワークフローはコードの再現性は保証しても、データの再現性は保証しないからである。ここでいうデータとはFASTQやBAMのような入力データだけでなく、リファレンスゲノムやBLASTデータベースのように、計算の途中で利用する参照データを意味する。
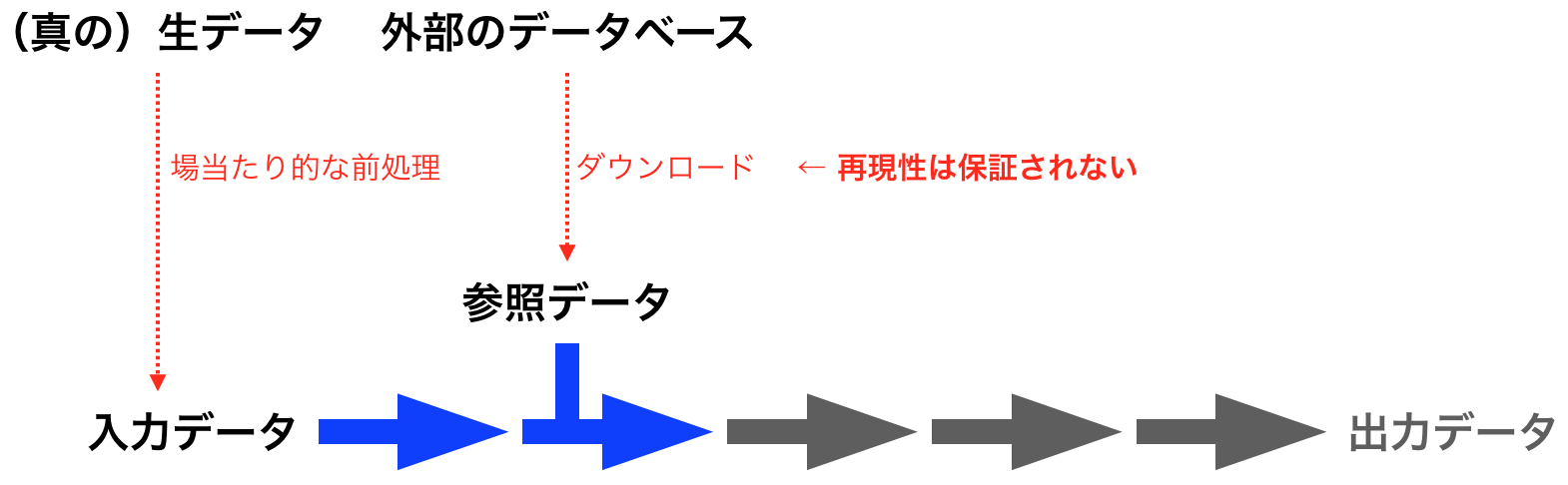
例えば、入力データに関しては、場当たり的にやるしか無い前処理の影響がある。多くのバイオのデータはノイジーで、アーティファクトを含んでいるため、前処理としてデータの一部を取り除く作業を行うことが多い。その作業の中で、空の行が含まれるFASTQや、極端に短くなったリード配列を作ってしまうこともあり、エラーを引き起こしたりする。つまり、そのワークフローや内部の解析ツールが想定していない状態のデータが入ることで動かなくなることは往々にしてある。これはユーザー、開発者のどちらが悪いというわけでは無いと思う。
また、参照データに関しても、外部のデータベースからダウンロードするため、ワークフロー外の出来事をワークフローは管理することができない。時には、サーバーが落ちていたり、突然廃止されたり、バージョンを固定せず日々更新していくタイプのデータベース(例: UniProtKB)や、データサイズが大きすぎてダウンロード中に接続が切れる場合も考えられる。また、ライセンスの関係で、アカウント登録をして、ログインしている状態のみでしかデータが取得できず、プログラマブルにデータを処理できないことさえある。
上記のような理由により、ワークフロー言語を使おうと、今後も何らかの理由で、計算がコケることは幾らでも起こりうる。実際に自分が開発したR/BioconductorのアノテーションパッケージであるMeSH.XXX.eg.dbやLRBase.XXX.eg.dbは、半年に一回更新しているが、必ずといっていいほど更新ワークフローが想定していないことが起きて、手作業で手直しをするステップが入っている。これは、上記のデータに関する不確かな部分が原因である。なお、データの再現は無理でも、ワークフローがコケた時に、原因究明がしやすくなるように、使ったツール・データのバージョン管理をしておくという観点でなら幾つかツールがある(例: Tximeta, AnnotationHub)。
ケース2: 試行錯誤
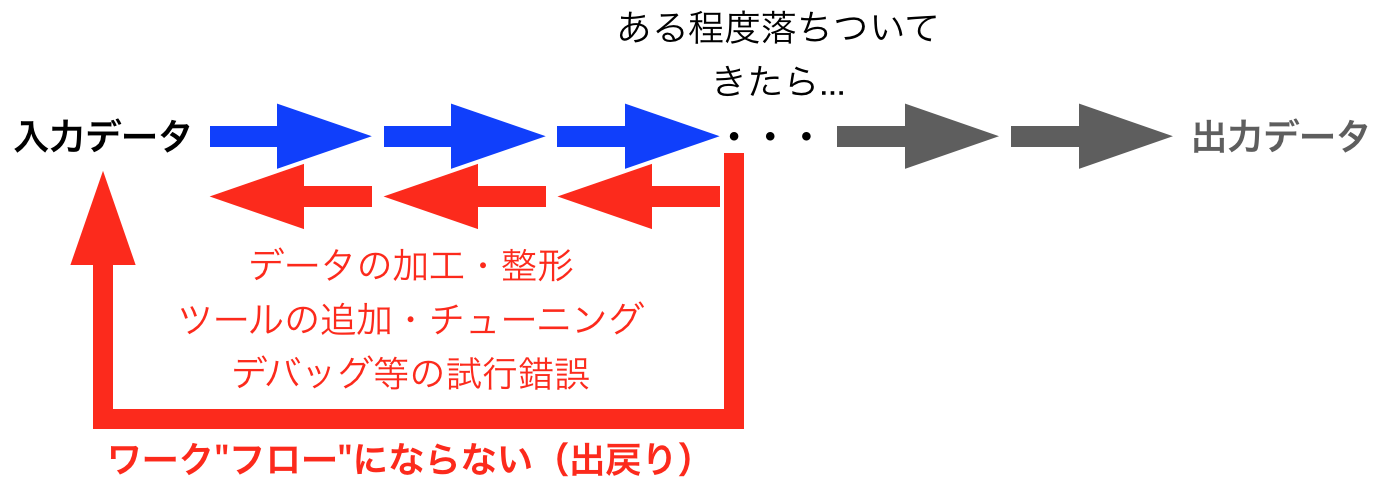
ワークフローによって、データから解析結果が簡単に得られるようになっても、実際のデータ解析は試行錯誤を伴う。生物由来のデータはノイズやアーティファクトが混入するため、データの品質管理の結果、フィルタリングをすることもあるし、また解析ツールのアルゴリズムのほとんどにはパラメーターがあり、その値によって解析結果は変わるのでチューニングしたいし、パラメーターの値次第ではコケることもある。また一見プログラムとしては正常に実行されているが、精度が極端に落ちることで、データやツールの使い方に不備があったというもっと厄介なケースもある。そのため自分としては、試行錯誤を経ていないワークフローから吐き出された解析結果を無条件に信用はできない。
例えば、1細胞オミックスの解析で言うならば、まずどのデータが何の細胞からわからない状態からスタートして、データをあらゆる側面で眺め回すことで、何の細胞なのかわかってくるという解析であり(cf. scTGIF)、そのデータに関する知識がアップデートされ、細胞型のラベルが更新されると、DEGやエンリッチメント解析などのラベルに依存した解析を再度やり直すことになる。
ワークフロー言語の売りの一つとして、タスク・ファイルの依存関係の有向無巡回グラフ(DAG)を考慮して、必要なところだけ上から順に実行してくれるという話しがあるが、そもそもそういう実行の仕方をしていない。途中まで試してみたコードをゴミ箱に捨てて、もう一度別のアプローチで最初からやり直す、みたいなこともよくやる。それでもRakefileなりで、ワークフロー風に書いているのは、単に後で見返しやすいようにメモとして残しているだけなので、手元のこれをワークフローとは呼びたくないと思っている。仮に上から順に実行するとしたら(多分やらないけど)、それはその研究のかなり最後の方だろう。
最近自分が書いたオンライン主成分分析のベンチマーク論文では20のPCAのツールを、4実データ、18人工データに対して実行して、どのPCAのツールが精度を落とすことなく、高速、低メモリに計算ができるかを評価した。この計算には、ラボのサーバ上で分散処理をしても3〜4週間はかかる。この論文においても、解析で利用したコードは一応ある程度きれいに整えた上で公開しているが、これをもう一回上から順にやる人間がいるのか?という疑問がある。それがめんどくさくて誰もやりたくないからこそ、やったことに価値があると思うのだが。こういった作業で、例えばCWLのような、考えることが多くて記述量が多いワークフロー言語を使っていた場合、折角書いたのに割りに合わない事態になるだろう。
試行錯誤した結果の残骸はワークフローの形をとるかもしれないし、試行錯誤を経てたどり着いたベストな(だと自分は思っている)ツールの使い方・手順や、それにより開発された新規手法・ツールなどは、ワークフローとして他人に使いやすい形態にするのは良いことかも知れない。しかし、試行錯誤の過程をワークフローが何か幸せにしてくれることはないと思う。また1ツール開発者的にも、あまり最初からワークフローを意識してコードを書くのは窮屈な気分になる。開発段階の汚い書き捨てコードを人様に見せたくないという心理も働くし、それらとの依存関係を最初から考慮してフローを構築したくないし、REPL(対話環境)でRを開きっぱなしにして関数を組み上げていく時間が長い中で、それをRのセッションを閉じて、毎回バッチ処理で再実行なんてしたくない(重いデータをロードしている時などは特に)。
ケース3: レポート
ワークフローを作ってそれで何がしたいのかということを突き詰めると、最後のアウトプットはおそらく人間が関係してくる。つまり、その解析結果を使って、論文、学会発表、社内会議、特許申請など、他の人間に何かを訴えかける活動をする事になる。ワークフローが解析結果を吐き出したとして、人間はその情報をありのままに理解することはできない。多次元データは一度次元圧縮して分布を俯瞰したくなるし、画像だけでなく、何がそこに書かれていて、何がわかったのかといった説明書きが加わって、初めて理解できる人は理解できるようになる。しかも、人間はすぐ疲れたり、飽きたりして、大量の画像・文章の処理を嫌がるので、その主張を支持するような厳選された情報だけを見せないといけない。そのため、手作業で画像の色やフォントサイズを調整したり、文章を何度も手直ししたりする。そういったことをワークフローはやってくれない。
 文芸的プログラミングの観点からすると、ワークフローにも、画像やら文章やらが関連づけられるべきなのだと思う。既存のワークフロー言語には、タスク間の依存関係(DAG)の可視化や、各タスクの計算時間をHTMLレポートにする機能などはあるが、各計算と画像や文章を結びつけるような機能は多分無い。そこから何が言えるのかという解釈(その計算時間が遅いのか速いのか、重いのか軽いのか、どこが注目すべき結果なのか、既存の知見と合うのかなど)は今後も自動化できないだろう。
文芸的プログラミングの観点からすると、ワークフローにも、画像やら文章やらが関連づけられるべきなのだと思う。既存のワークフロー言語には、タスク間の依存関係(DAG)の可視化や、各タスクの計算時間をHTMLレポートにする機能などはあるが、各計算と画像や文章を結びつけるような機能は多分無い。そこから何が言えるのかという解釈(その計算時間が遅いのか速いのか、重いのか軽いのか、どこが注目すべき結果なのか、既存の知見と合うのかなど)は今後も自動化できないだろう。
最近PapermillでJupyter Notebookをバッチ処理するやり方を知ったので、これが多少はこの作業を緩和してくれるのではと期待している。またF1000誌のBioconductorゲートウェイでは、Bioconductorのワークフローパッケージのvignetteをそのまま論文としても受け付けてくれるので、ワークフロー開発 = 論文1報扱いという時代がきて、それならワークフローを丁寧に書いても良いかという気になるかも知れない。
まとめ
上記のように、ワークフローは万能ではないし、ワークフローになっていたとしても、いつでもどこでも動くわけではない。そのため、ワークフローを書くということは、「できるだけ再現良く動くようにする努力・試み」ぐらいのニュアンスのほうが正確だと思う。または、現状うまくいってないからこそ、研究として取り組んでいる人がいると考えるべきなのかもしれない。ワークフローを書くこと自体もそれなりに労力はかかるし、それに見合うかよく考えて、自分に必要なら使えば良いと思う。自分は試行錯誤の中でワークフローを書くメリットは感じなかった。
ワークフローのユーザーに言いたいのは、既存のツールやワークフローを使えるようになったからといって、何かを達成した気になってはいけないと思う。それらツールもワークフローもあなたが作ったものではないし、それらを単に動かせるようになっただけでは、データ解析としてはまだ入り口ぐらいである。もっとデータを血眼になって眺めて、早くそこから生物学的な知見を得て、人類を前に進めて欲しい。IT技術は流行り廃りある中で、自分の仕事に便利に使えるなら取り入れれば良くて、決して手段と目的を履き違えてはいけない。
また、ワークフローの開発者に関しては、その業界のデファクトスタンダード的なツールを公開しただけで成果としている印象があるが、そのアプローチで幸せになるのは、コードを書きたくないWet研究者であったり、大量の共同研究先を抱えていて、完璧なクオリティーとまではいかなくても、とにかく大量に解析をさばくことが業務のDry研究者などでは無いだろうか。自分は今後もできあいのワークフローだけで実データ解析を行うことは少ないと思われるため、ワークフローの恩恵は受けられないと感じている。