はじめに
Microsoft Genomicsのクイックスタート をやってみた際に少し分かりにくいポイント・詰まりやすいポイントがあったので、一通りの使い方を自分なりにまとめました。
Microsoft Genomics とは
DNAシーケンサーで得られたFASTAQファイル、BAMファイル等を対象に、クラウド上でBurrows-Wheeler Aligner (BWA)やScalable Nucleotide Alignment Program (SNAP)による配列のマッピング、 Genome Analysis Toolkit (GATK) による変異検出等のゲノム解析を行ってくれるサービス
シーケンサーで得られた配列データをAzure Storage上に置いたまま解析ができ、また解析ツールの環境構築やコンピューターリソースの管理を行う手間がいらず便利
参考:
Microsoft Genomics
マイクロソフト、ゲノム研究用クラウドツールの一般提供開始を発表
前提条件
- Azureアカウントと有効なサブスクリプション
- Python 2.7.12以上が入った環境
※Microsoft GenomicsのPythonクライアントはPython 3と互換性がありません (2020.5.24現在)
pyenvを使用して複数のPythonバージョンを切り分けて使いたい場合は下記記事を参照してください。macOS, Linux, Windows (WSL)環境に対応しています。
HomebrewのインストールからpyenvでPythonのAnaconda環境構築までメモ
Microsoft Genomicsのセットアップ
アカウントの作成
Azure Portalで「Genomics」と検索してGenomicsアカウントの作成ページへ行きます。
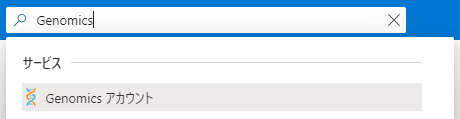
必要事項を入力し、作成を行います。
構成ファイルのダウンロード
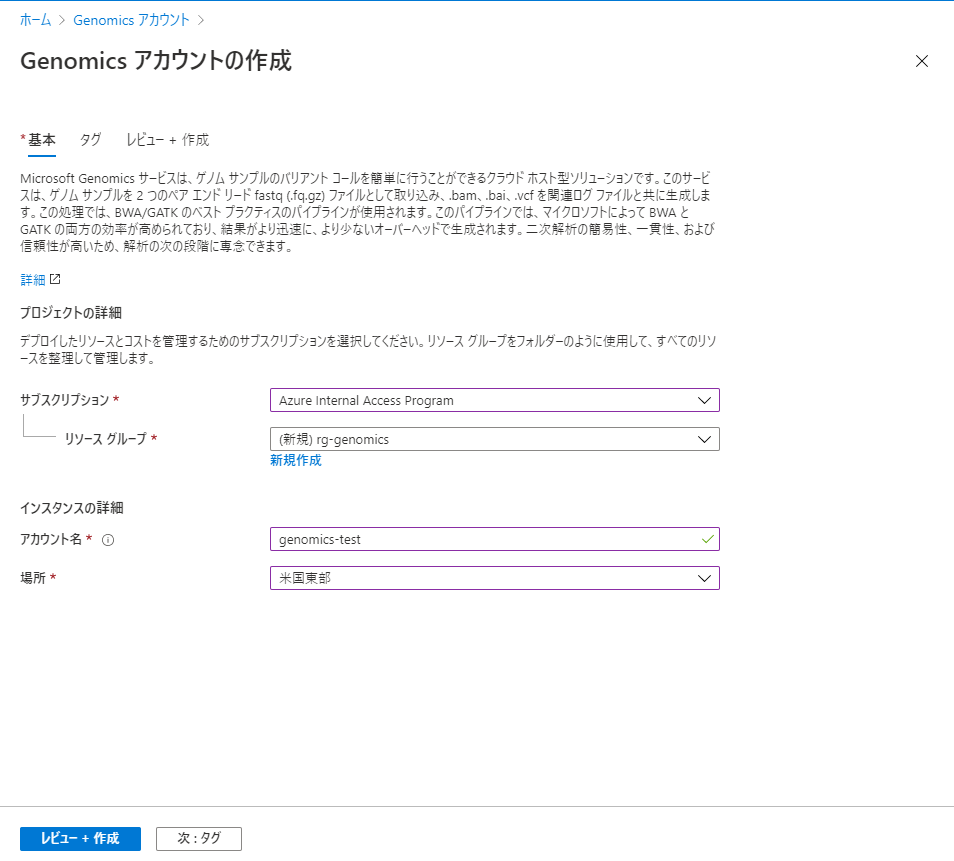
再度「Genomics」と検索し、Genomics アカウントのページへ行きます。
先ほど作成した Genomics アカウントを選択して、「アクセス キー」に移動し、構成ファイルをダウンロードしておきます。

config.txt としてダウンロードされます。
Pythonクライアントのインストール
Python 2.7.12以上がインストールされている環境でMicrosoft GenomicsのPython クライアントをインストールします。
$ pip install msgen
※もし特に何もいじっていないLinux, WSL環境でインストールしたい場合は下記コマンドを実行してみてください (管理者権限が必要です)
sudo apt-get install -y build-essential libssl-dev libffi-dev libpython-dev python-dev python-pip
sudo pip install --upgrade --no-deps msgen
sudo pip install msgen
以下のコマンドでMicrosoft GenomicsのPythonクライアントの動作を確認できます。
$ msgen list -f ./config.txt
(config.txtへのファイルパスを指定してください)
無事にインストールされていて、config.txtファイルが認識されていれば次のようになります。

ストレージの準備
ストレージアカウントの作成
入力ファイルと出力ファイルを格納するAzure Storageアカウントを作成します。
Azure Portalから、「ストレージアカウント」と検索し作成を行います。
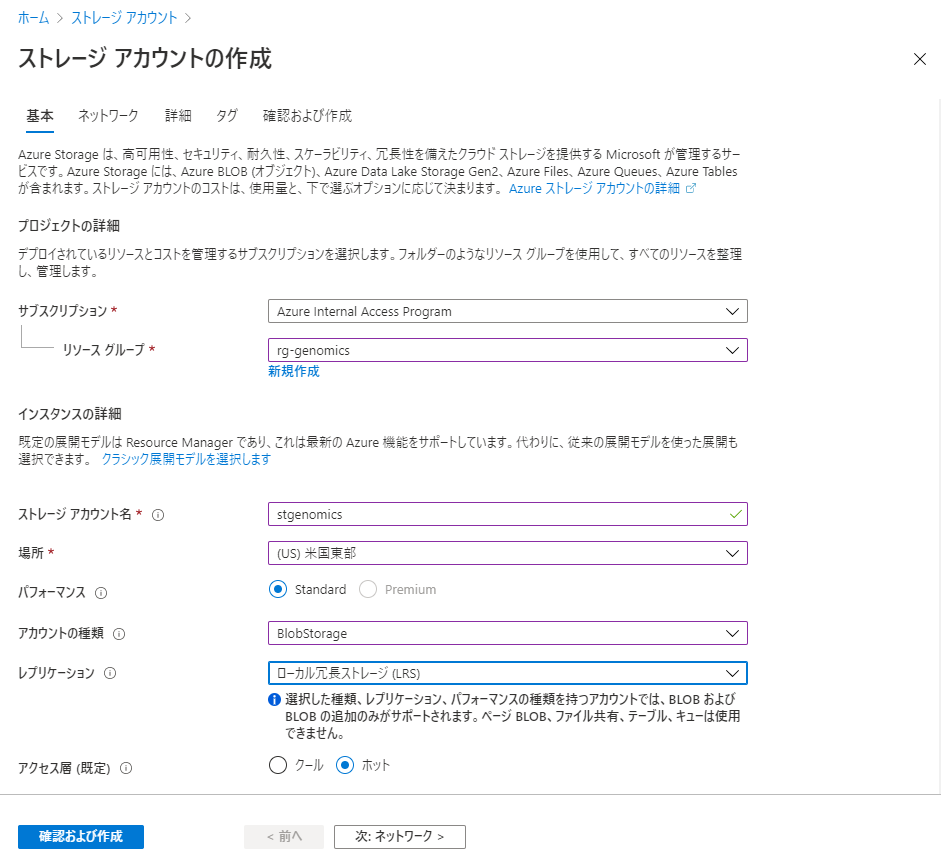
展開するリージョン (場所)はGenomicsアカウントと同じ場所にすることでデータ移動料金や待ち時間を短縮します。
「確認および作成」を選択してストレージアカウントを作成します。
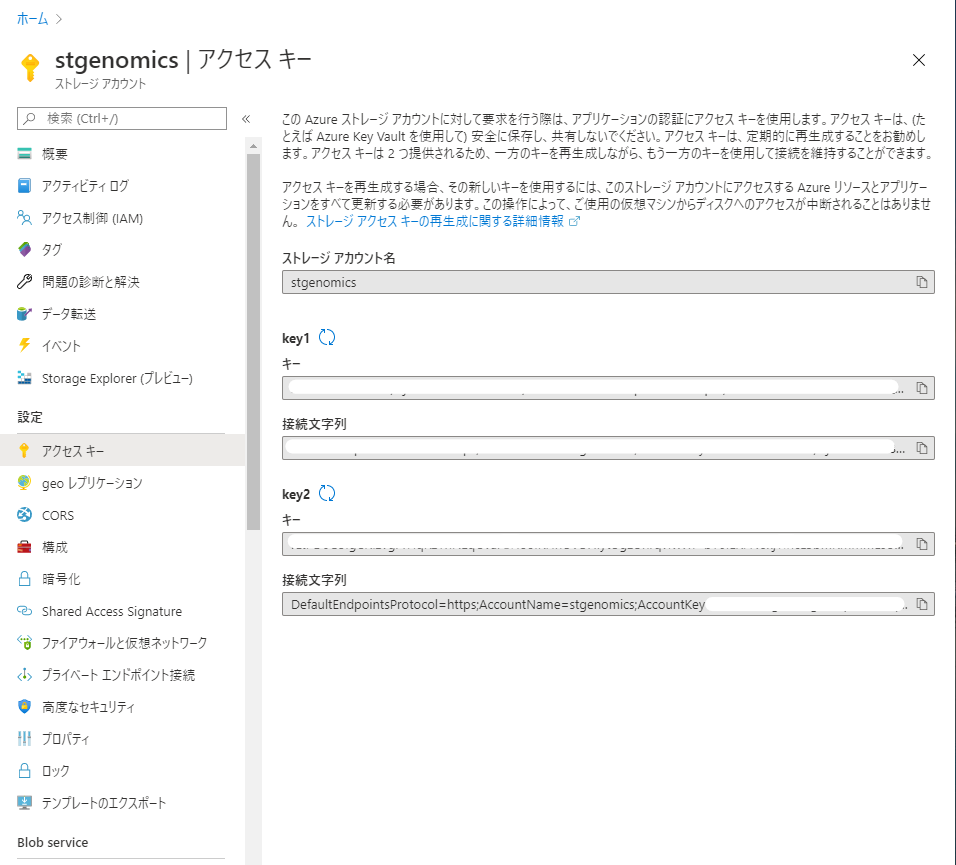
アカウントを作成したら、ストレージアカウント画面左側より「アクセス キー」を選択し、アクセスキー情報を保存しておきます (ブラウザの別タブでアクセスキーのページを開いておくと便利です)
Blob コンテナーの作成
ストレージアカウントのページへいき、左側メニューからコンテナーを選択します。

上部の「+コンテナー」を選択し、新しいコンテナーを作成します。
入力ファイル保管用と出力ファイル保管用に二つのコンテナーを作成します。

ここではそれぞれinput-files, output-files という名前にしています。

入力データのストレージアカウントへのアップロード
入力データの準備
今回はクイックスタートでサンプルデータとして提供されている以下のデータを使用します。
https://msgensampledata.blob.core.windows.net/small/chr21_1.fq.gz
https://msgensampledata.blob.core.windows.net/small/chr21_2.fq.gz
名前から察するにヒトの21番染色体のシーケンスデータですね。
これらをダウンロードして保存しておきます(容量大きいので注意)
Azure Portal上でのアップロード
ストレージアカウントへのアップロード方法は多数存在 (Azure Storage Explorer, AzCopyコマンド, etc.)しますが、今回はシンプルにPortal上でのアップロードを行います。
※コマンドライン環境下でのアップロードを行いたい場合はAzCopyコマンド等の使用を検討してください。
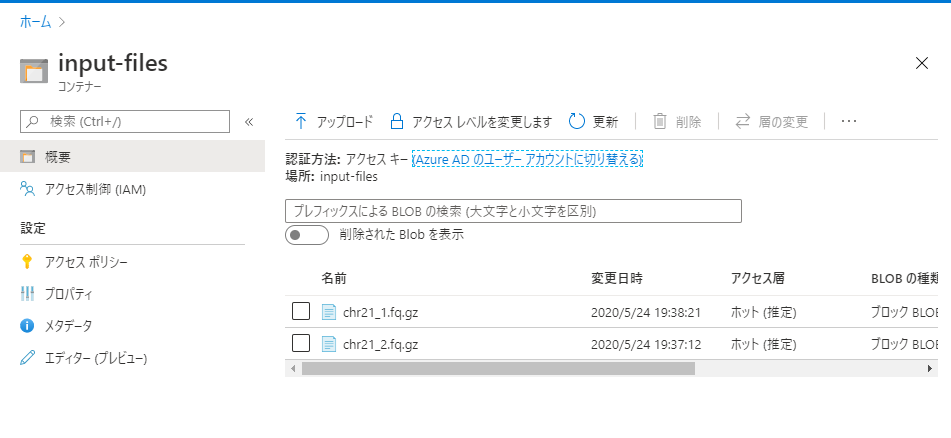
先ほど作成したinput-files ストレージコンテナーの管理画面へ行きます。

画面中央上部の「アップロード」を選択し、先ほどダウンロードした
chr21_1.fq.gz, chr21_2.fq.gz のファイルをそれぞれ選択してアップロードします。

両方ともしっかりアップロードします。

アップロードが完了しました。
Microsoft Genomics ワークフローを実行する
configファイルの編集
冒頭でダウンロードしたconfig.txt ファイルを開きます。(筆者はVSCodeで開いていますがメモ帳等でも結構です)

デフォルトではヒトリファレンスゲノムはhg19を使用するようになっています。
GATKはb37, hg38, hg38 (alt 分析なし), hg19の4つのヒトリファレンスゲノムに対応しているようです。
参考:ヒトのリファレンスゲノムについて, (日本語解説記事)
※もしGATK4を実行したい場合は、process_nameパラメーターをgatk4に設定します。
画像下部の箇所にストレージキーの情報を入力します。

ファイルを保存します。
ワークフローの送信
Microsoft Genomics Pythonクライアントを使用し、以下のフォーマットでワークフローを送信します。
$ msgen submit -f [full path to your config file] -b1 [name of your first paired end read] -b2 [name of your second paired end read]
筆者の環境では以下のようになりました。

Message: Successfully submitted と表示されていればワークフローの送信は成功です。
以下のコマンドでワークフローの状態を確認できます。
$ msgen list -f ./config.txt
このように表示されます。

適宜状態を確認し、以下のようなStatusになったら実行完了です。

ワークフローの実行が完了すると、先ほど作成したAzure Storage アカウントの出力コンテナー内に出力ファイルが作成されます。

出力先にあるファイルたち
- logs.zip:ツールの実行ログたち
- BAMファイル:シーケンサーから出力されたリードをリファレンス配列にマッピングした結果
- .bam.baiファイル:BAMのインデックス情報
- VCF (variant call format)ファイル:今回のヒト21番染色体のシーケンスデータをヒトリファレンスゲノムにマッピングした結果得られた、ゲノムの変異情報 (リファレンスゲノムと異なる部分が抽出されている)が保存されたファイル
詳しい情報:ファイルフォーマットについて
Next Steps
得られたBAMやVCFファイルをもとに可視化を行えます。
例えばこちらを参照
bamとvcfの可視化分析ツール bam.iobio.ioとvcf.iobio.io
分かりやすくかっこいい図が出せます。

(上記記事より転載)