scRNA-seq では、1細胞ごとに数千から数万遺伝子の発現量を扱います。つまり、各細胞は「遺伝子数次元のベクトル」として表されます。PBMC3k のような比較的小さなデータでも、解析対象は 2,700 細胞 × 13,714 遺伝子という高次元データです。
この高次元データをそのまま眺めることはできません。そのため、PCA、t-SNE、UMAP のような次元削減を使って、細胞間の構造を探索・可視化します。
ただし、最初に強調しておきたいことがあります。
- PCA、t-SNE、UMAP は「クラスタリングの正解」ではありません。
- UMAP や t-SNE で離れて見えるからといって、必ずしも生物学的に遠いとは限りません。
- クラスタ ID は便宜上のラベルであり、真の細胞型ラベルではありません。
- 可視化結果だけで生物学的結論を確定せず、既知マーカー、差次的発現、実験条件、外部知識と合わせて解釈します。
本記事では、10x Genomics が公開している PBMC3k データを使い、PCA、t-SNE、UMAP が何を最適化しているのかを数式から確認しながら、Seurat で実際に比較します。
目次
- 再現用コード一式
- 使用データ:10x Genomics PBMC3k
- 前処理:なぜこの順番で処理するのか
- PCA:線形代数としての次元削減
- PC数を変えると何が変わるか
- t-SNE:近傍確率を合わせる非線形可視化
- UMAP:近傍グラフの構造を保つ
- 定量評価:見た目だけで判断しない
- 実務上のまとめ
- 参考文献・公式資料
再現用コード一式
再現用コード、図表、GIF は以下の GitHub リポジトリにまとめています。
本記事の再現コードは以下の構成です。
.
├── article/index.md
├── scripts/
│ ├── 00_setup.R
│ ├── 01_download_pbmc3k.R
│ ├── 02_seurat_preprocess.R
│ ├── 03_pca_analysis.R
│ ├── 04_tsne_umap_parameter_sweep.R
│ ├── 05_metrics.R
│ └── 06_make_gifs.R
├── figures/
├── gifs/
├── data/
├── results/
├── references.bib
└── README.md
実行順序は README にまとめています。データ本体は Git 管理対象にせず、scripts/01_download_pbmc3k.R で 10x Genomics から再取得します。
使用データ:10x Genomics PBMC3k
使用するデータは、10x Genomics の 3k PBMCs from a Healthy Donor です。
| 項目 | 内容 |
|---|---|
| データ | 3k PBMCs from a Healthy Donor |
| 由来 | 健常ドナー由来の peripheral blood mononuclear cells |
| 細胞数 | 2,700 cells detected |
| シーケンサー | Illumina NextSeq 500 |
| リード数 | 約 69,000 reads/cell |
| 解析 | Cell Ranger 1.1.0 |
| ライセンス | Creative Commons Attribution 4.0 International (CC BY 4.0) |
| 公開元 | 10x Genomics |
Seurat 公式の PBMC3k guided clustering tutorial でも、この 10x Genomics 公開データが使われています。
ダウンロードは以下のスクリプトで行います。
Rscript scripts/01_download_pbmc3k.R
前処理:なぜこの順番で処理するのか
Seurat の標準的な流れに沿って、以下を実行します。
pbmc.data <- Read10X(data.dir = "data/filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(
counts = pbmc.data,
project = "pbmc3k",
min.cells = 3,
min.features = 200
)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
pbmc <- ScaleData(pbmc, features = rownames(pbmc))
pbmc <- RunPCA(pbmc, features = VariableFeatures(pbmc), npcs = 50)
各ステップの意味は次のとおりです。
| ステップ | 目的 |
|---|---|
Read10X() |
Cell Ranger 出力の UMI count matrix を読み込む |
CreateSeuratObject() |
カウント行列とメタデータを Seurat オブジェクトとして保持する |
PercentageFeatureSet() |
ミトコンドリア遺伝子割合を QC 指標として計算する |
| QC filtering | 低品質細胞や multiplet 疑いの細胞を除外する |
NormalizeData() |
細胞ごとの総 UMI 数の違いを補正し、log 変換する |
FindVariableFeatures() |
細胞間変動が大きい遺伝子を抽出する |
ScaleData() |
遺伝子ごとに平均0・分散1へ変換し、PCAに備える |
RunPCA() |
高次元発現行列を低次元の主成分に圧縮する |

この図で見るべきこと:nFeature_RNA、nCount_RNA、percent.mt の分布です。極端に低い nFeature_RNA は空ドロップレットや低品質細胞、極端に高い nFeature_RNA は multiplet の可能性があります。
この図だけでは言えないこと:細胞型の正体は決まりません。QC は解析対象として妥当な細胞を残すための入口です。
PCA:線形代数としての次元削減
発現行列の定義
細胞数を $n$、遺伝子数を $p$ とします。scRNA-seq の発現行列を
$$
X \in \mathbb{R}^{n \times p}
$$
と書きます。行が細胞、列が遺伝子です。
Seurat では通常、正規化後に高変動遺伝子を選び、ScaleData() によって遺伝子ごとに中心化・標準化した行列に対して PCA を行います。
中心化・標準化後の行列を改めて $X$ とすると、各遺伝子列は平均0、分散1に近い状態になります。
分散共分散行列
PCA は、分散が最大になる方向を順番に探す方法です。標準化済み行列 $X$ に対して、分散共分散行列は
$$
S = \frac{1}{n - 1} X^\top X
$$
です。
ここで $S$ は $p \times p$ の行列で、遺伝子間の共変動を表します。
固有値問題
PCA は次の固有値問題を解きます。
$$
S v_k = \lambda_k v_k
$$
$v_k$ は第 $k$ 主成分の方向、$\lambda_k$ はその方向に沿った分散です。$\lambda_k$ が大きいほど、その主成分はデータの分散をよく説明します。
主成分スコア
各細胞を第 $k$ 主成分軸へ射影した値が主成分スコアです。
$$
z_k = X v_k
$$
細胞を PC1 と PC2 で散布図にすると、各細胞の $z_1, z_2$ を描いていることになります。
loading の意味
loading は、各遺伝子が主成分方向にどれだけ寄与しているかを表します。PC1 の loading が大きい遺伝子は、PC1 方向の細胞間差を作る遺伝子です。
PBMC3k では、T細胞、B細胞、単球、NK細胞、血小板関連遺伝子などが上位 loading に現れることがあります。
explained variance と Elbow plot
第 $k$ 主成分の explained variance ratio は、おおまかに
$$
\frac{\lambda_k}{\sum_j \lambda_j}
$$
です。Elbow plot は、PC番号に対して explained variance を並べた図です。急に落ち込みが緩やかになる位置を「情報量とノイズの境界」の目安として見ます。

この図で見るべきこと:PCAだけでも大まかな細胞集団の分離が見えるか、特定のクラスタがPC軸に沿って分かれているかです。
この図だけでは言えないこと:クラスタが細胞型として確定したとは言えません。loading とマーカー遺伝子を確認する必要があります。
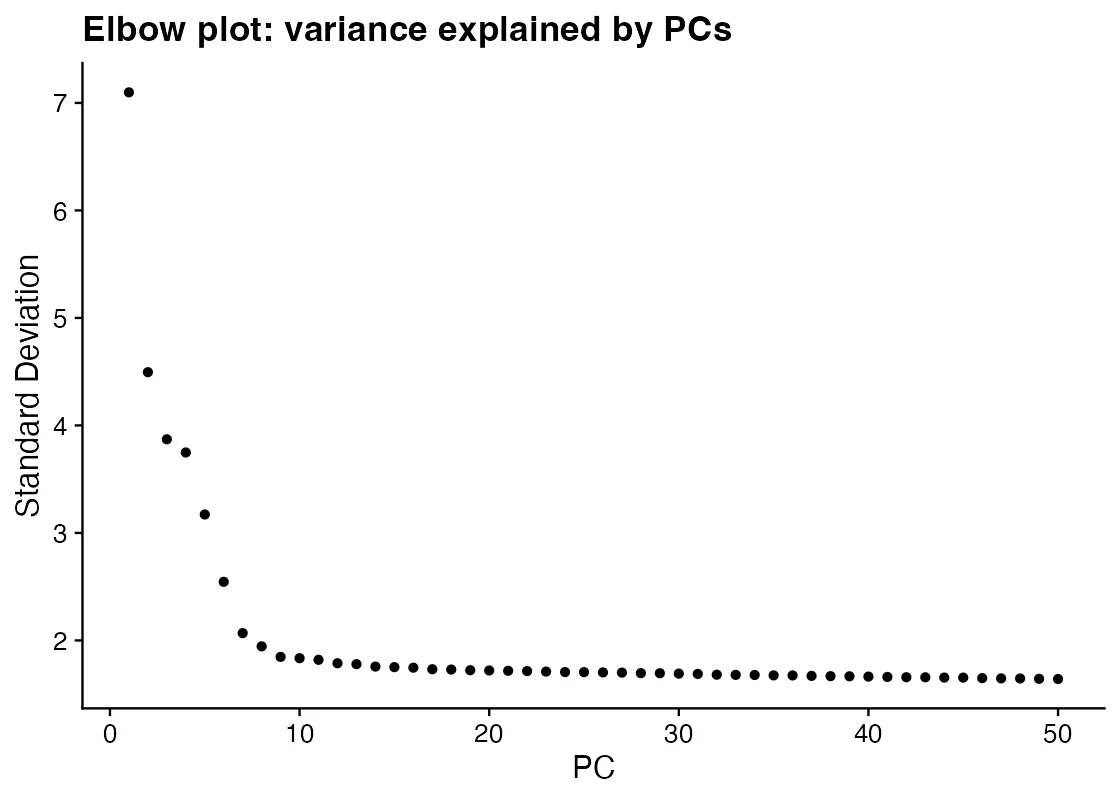
この図で見るべきこと:どのPCまでを下流解析に使うかの目安です。Seurat公式チュートリアルでは、PBMC3kでPC 7から12程度が一つの候補として説明されています。
この図だけでは言えないこと:最適なPC数が一意に決まるわけではありません。希少細胞集団が後ろのPCに出ることもあります。

この図で見るべきこと:主成分がどの遺伝子群に支えられているかです。loading を見ることで、PCAが単なる数学的圧縮ではなく、生物学的シグナルの候補を含むことがわかります。

この図で見るべきこと:PCごとに高スコア・低スコアの細胞と遺伝子がどのように対応しているかです。
PCA の直感 GIF
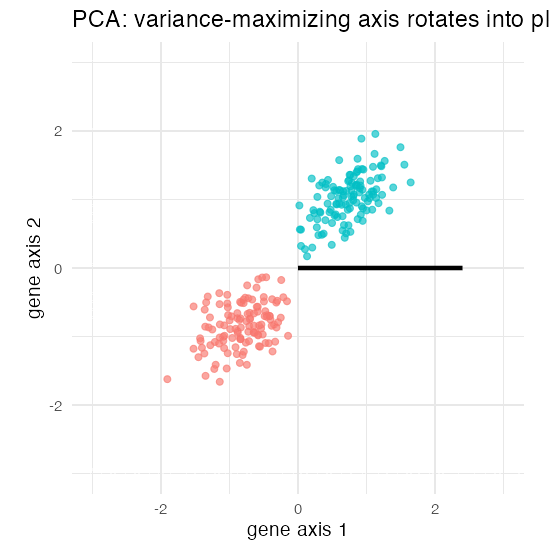
このGIFで見るべきこと:PCAは、点群の分散が最も大きい方向へ軸を回転させる操作だという直感です。
このGIFだけでは言えないこと:実際のscRNA-seqは非線形構造やドロップアウトを含むため、PCAだけですべての構造を説明できるわけではありません。
PC数を変えると何が変わるか
本記事では以下のPC数で下流解析を比較します。
dims = 1:5dims = 1:10dims = 1:20dims = 1:30dims = 1:50
各PC数について、UMAP、t-SNE、Seurat clustering、cluster数、実行時間、trustworthiness、kNN preservation を出力します。
PC数が少なすぎると、希少細胞集団や弱い生物学的シグナルが落ちる可能性があります。一方で、PC数を増やしすぎると、技術ノイズや細胞周期などの不要な変動を拾う可能性があります。
Seurat公式チュートリアルでも、PBMC3kでは 5 PCs だけでは結果に悪影響が出ることがあり、10、15、50など複数のPC数を試すことが推奨されています。
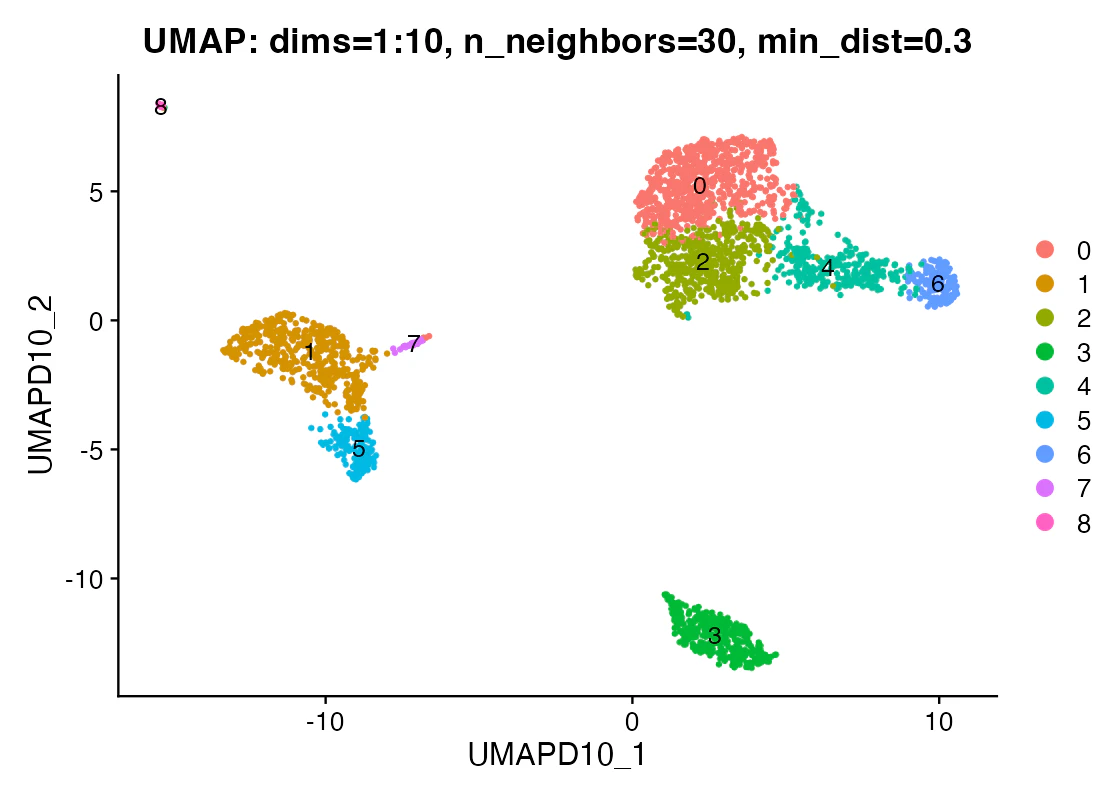
この図で見るべきこと:PC数を固定したとき、UMAP上で主要なPBMC集団らしい構造が見えるかです。
この図だけでは言えないこと:クラスタ間距離の大きさを、そのまま発現差の大きさとして読んではいけません。
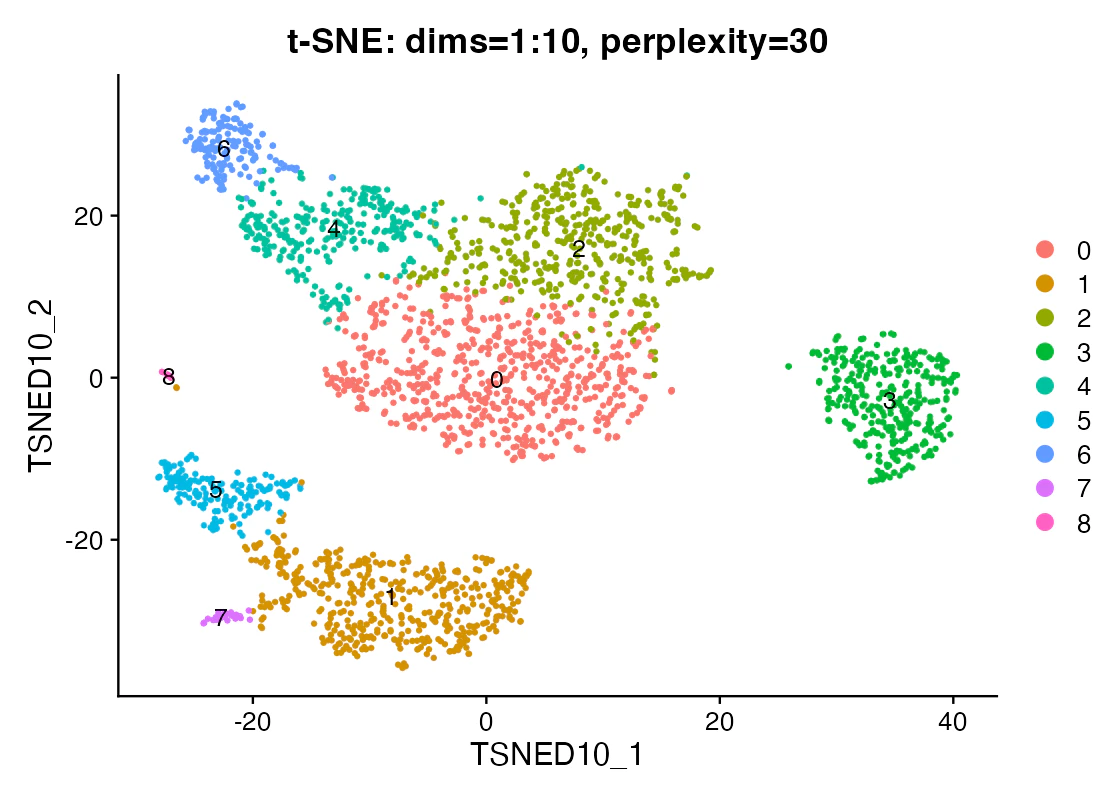
この図で見るべきこと:局所的に近い細胞がまとまりやすいかです。
この図だけでは言えないこと:t-SNE上のクラスタサイズやクラスタ間距離は、元空間の密度や距離を忠実に表しているとは限りません。
t-SNE:近傍確率を合わせる非線形可視化
t-SNE は、高次元空間と低次元空間の「近さの確率分布」を近づける方法です。
高次元空間での条件付き確率
高次元空間の点 $x_i$ に対して、点 $x_j$ が近傍として選ばれる条件付き確率を
$$
p_{j|i} =
\frac{
\exp\left(-|x_i - x_j|^2 / 2\sigma_i^2\right)
}{
\sum_{k \ne i}
\exp\left(-|x_i - x_k|^2 / 2\sigma_i^2\right)
}
$$
と定義します。
$\sigma_i$ は点 $i$ ごとに異なり、perplexity に合うように調整されます。
perplexity の直感
perplexity は「各点が何個くらいの近傍を意識するか」に近いパラメータです。小さい perplexity は非常に局所的な構造を強調し、大きい perplexity はより広い近傍を見ます。
ただし、perplexity は単純な近傍数そのものではありません。確率分布のエントロピーに基づく有効近傍数です。
低次元空間での確率
低次元座標を $y_i$ とします。t-SNE では低次元空間で Student-t 分布を使って
$$
q_{ij} =
\frac{
(1 + |y_i - y_j|^2)^{-1}
}{
\sum_{k \ne l}(1 + |y_k - y_l|^2)^{-1}
}
$$
と定義します。
Student-t 分布を使う理由は、低次元空間で中程度に離れた点を強く押しつぶしすぎないためです。これにより、いわゆる crowding problem を緩和します。
KL divergence の最小化
t-SNE は
$$
C = \sum_{i \ne j} p_{ij} \log \frac{p_{ij}}{q_{ij}}
$$
を最小化します。
これは $P$ で近い点が $Q$ でも近くなるように低次元座標 $y_i$ を更新する、という意味です。最適化は勾配降下法で行われます。
t-SNE の注意点
t-SNE は局所構造を見やすくする強力な可視化手法ですが、以下には注意が必要です。
- cluster size を細胞数や密度としてそのまま解釈しない
- cluster間距離を生物学的距離としてそのまま解釈しない
- perplexity や乱数 seed によって見え方が変わる
- 可視化結果からクラスタ数を決め打ちしない
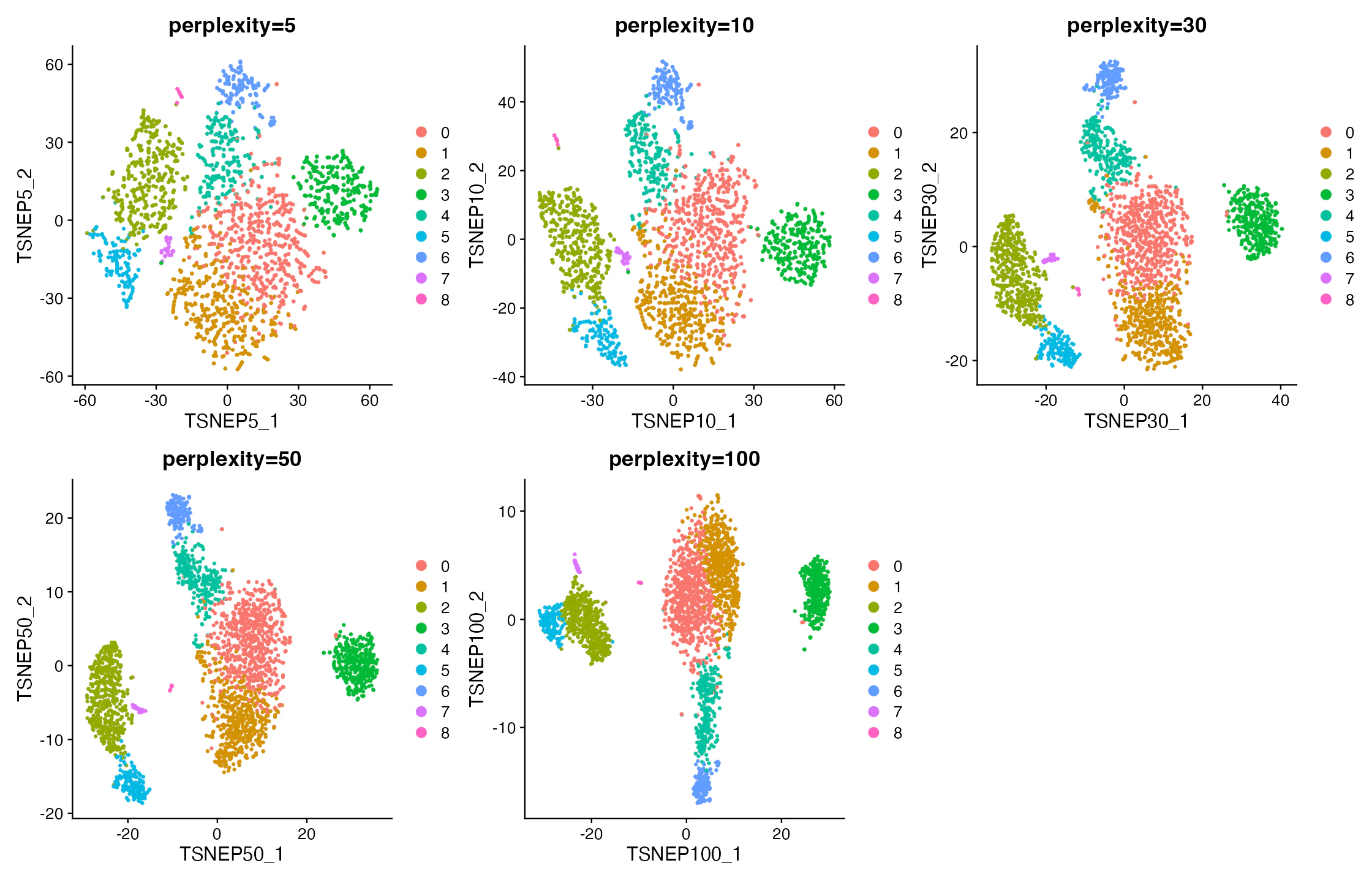
この図で見るべきこと:perplexity を小さくすると局所的なまとまりが強調され、大きくするとより広い近傍構造が反映されやすくなる点です。
この図だけでは言えないこと:perplexity 100 が標準的に良い、という意味ではありません。PBMC3k規模では極端な比較例として扱います。
このGIFで見るべきこと:t-SNE は最初からクラスタを知っているのではなく、低次元座標を反復的に更新して近傍確率を合わせにいく、という点です。
UMAP:近傍グラフの構造を保つ
UMAP は、k近傍グラフに基づいて高次元空間の局所構造を表し、それに対応する低次元配置を求める方法です。
k近傍グラフ
各点 $x_i$ について、近い点を $k$ 個探します。この $k$ に対応する主要パラメータが n_neighbors です。
n_neighbors が小さいと局所構造を強く見ます。大きいと、より広い構造を反映しやすくなります。
局所距離スケールと fuzzy simplicial set
UMAP では、各点の近傍距離に応じて局所的な距離スケールを推定します。直感的には、密な領域と疎な領域で「近い」の基準を調整します。
高次元空間で点 $i$ と $j$ がどれくらい近傍関係にあるかを $\mu_{ij}$ とします。これは fuzzy simplicial set と呼ばれる、近傍関係の重み付きグラフのように考えると理解しやすいです。
低次元空間での近傍関係
低次元空間の近傍関係は、代表的には
$$
\nu_{ij} = \frac{1}{1 + a|y_i - y_j|^{2b}}
$$
のような形で表されます。
$a$ と $b$ は min_dist などから決まる曲線の形を制御するパラメータです。
cross entropy の最小化
UMAP は、高次元の近傍関係 $\mu_{ij}$ と低次元の近傍関係 $\nu_{ij}$ の差を cross entropy として評価し、それを小さくするように $y_i$ を更新します。
直感的には、元空間で近い点は低次元でも近く、元空間で近くない点は必要以上に近づきすぎないように配置します。
UMAP の主要パラメータ
| パラメータ | 意味 |
|---|---|
n_neighbors |
各点がどの程度広い近傍を見るか |
min_dist |
低次元空間で点をどれくらい密に詰めてよいか |
metric |
元空間で距離をどう測るか |
本記事では、PCA空間上の近傍探索に対して metric = "cosine" を基本にします。ユークリッド距離も一般的ですが、cosine は方向の類似性を重視するため、高次元・疎な発現データや正規化済み特徴量で使われることがあります。実務では、Seuratの既定値や解析目的と合わせて比較します。

この図で見るべきこと:n_neighbors を小さくすると局所構造が強調され、大きくすると全体の連続性を見やすくなることです。
この図だけでは言えないこと:細かく分かれた見た目が、必ずしも生物学的に正しいクラスタ分割とは限りません。

この図で見るべきこと:min_dist が小さいほどクラスタが詰まり、min_dist が大きいほど点群が広がります。
この図だけでは言えないこと:密に詰まったクラスタが、必ず強い生物学的まとまりを意味するわけではありません。

この図で見るべきこと:n_neighbors と min_dist は別々に効きます。前者は近傍スケール、後者は低次元での詰まり方を主に変えます。

このGIFで見るべきこと:UMAPでは近傍グラフの情報が低次元配置に反映され、近傍の見方を変えると局所構造と大域構造のバランスが変わる、という直感です。

このGIFで見るべきこと:同じPBMC3kでも min_dist を変えると見た目の詰まり方が変わることです。
このGIFだけでは言えないこと:これは同じ初期配置から滑らかに変形する「最適化の軌跡」ではありません。各 min_dist で独立に最適化した UMAP 配置を順に表示しているため、フレーム間の絶対的な移動距離や回転は解釈しません。GIFの見た目だけで「最適な min_dist」は決められません。マーカー遺伝子、クラスタ安定性、解析目的と合わせて選びます。
定量評価:見た目だけで判断しない
本記事のスクリプトでは、以下を計算します。
- runtime
- cluster数
- trustworthiness
- kNN preservation
- ARI / NMI
- 既知マーカーに基づく簡易確認
trustworthiness は、低次元で近くなった点が元空間でも近いかを見る指標です。scikit-learn の trustworthiness と同じ考え方で、0から1の値を取り、1に近いほど近傍関係の破綻が少ないことを意味します。
kNN preservation は、高次元PCA空間のk近傍が低次元空間でもどれくらい保存されるかを直接数えます。
ARI / NMI はクラスタラベル同士の一致度です。ただし、本記事では Seurat cluster を比較対象にしているため、これは真の正解ラベルに対する精度ではありません。あくまで、PC数や埋め込み条件を変えたときにクラスタリング結果がどれくらい変わるかを見る補助指標です。
PBMCらしい構造の確認には、以下のような既知マーカーを使います。
| 細胞集団 | 例 |
|---|---|
| T細胞 |
CD3D, CD3E, IL7R
|
| B細胞 |
MS4A1, CD79A
|
| NK細胞 |
NKG7, GNLY
|
| 単球 |
LYZ, S100A8, S100A9, FCGR3A, MS4A7
|
| 樹状細胞 |
FCER1A, CST3
|
| 血小板 |
PPBP, PF4
|
ここでも注意すべき点は、マーカーによる簡易注釈は最終的な細胞型アノテーションではないということです。研究用途では、より体系的な marker validation や参照データへの mapping を行います。
実務上のまとめ
PCA、t-SNE、UMAP はそれぞれ役割が異なります。
| 手法 | 主な役割 | 強み | 注意点 |
|---|---|---|---|
| PCA | 線形圧縮、ノイズ低減、下流解析の入力 | 解釈しやすい、loadingを見られる | 非線形構造は表現しにくい |
| t-SNE | 局所構造の可視化 | 局所クラスタが見やすい | 距離・サイズ・大域構造の解釈に注意 |
| UMAP | 近傍グラフ構造の可視化 | 局所と大域のバランスを取りやすい | パラメータで見た目が大きく変わる |
実務では、まず PCA でノイズを抑えた表現を作り、その PCA 空間を使って clustering、UMAP、t-SNE を行うことが多いです。
そのうえで、以下を確認します。
- PC数を変えても主要な構造が大きく崩れないか
- UMAP / t-SNE のパラメータを変えても解釈が過度に変わらないか
- cluster ID を細胞型ラベルと混同していないか
- 既知マーカーでPBMCらしい構造が確認できるか
- 可視化から言えることと言えないことを分けているか
「UMAPがきれいだから正しい」ではなく、「どの空間で、どの近傍関係を、どの目的関数で保とうとしているのか」を理解して使うことが重要です。
参考文献・公式資料
| # | 資料 | 関連 |
|---|---|---|
| 1 | 10x Genomics: 3k PBMCs from a Healthy Donor | PBMC3k データ本体、細胞数、ライセンス |
| 2 | Seurat Guided Clustering Tutorial (PBMC 3k) | Seurat公式ワークフロー |
| 3 | Seurat RunTSNE documentation | Seuratでのt-SNE実行 |
| 4 | Seurat RunUMAP documentation | SeuratでのUMAP実行 |
| 5 | van der Maaten L, Hinton G. Visualizing Data using t-SNE. JMLR. 2008. | t-SNE原著 |
| 6 | Wattenberg M, Viégas F, Johnson I. How to Use t-SNE Effectively. Distill. 2016. | t-SNEの解釈上の注意 |
| 7 | Kobak D, Berens P. The art of using t-SNE for single-cell transcriptomics. Nature Communications. 2019. | scRNA-seqでのt-SNE実務 |
| 8 | McInnes L, Healy J, Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. 2018. | UMAP原著 |
| 9 | umap-learn documentation: Basic UMAP Parameters |
n_neighbors, min_dist, metric の解説 |
| 10 | Single-cell best practices: Dimensionality reduction | scRNA-seq次元削減の実務的整理 |
| 11 | scikit-learn trustworthiness documentation | trustworthiness 指標 |