1. はじめに
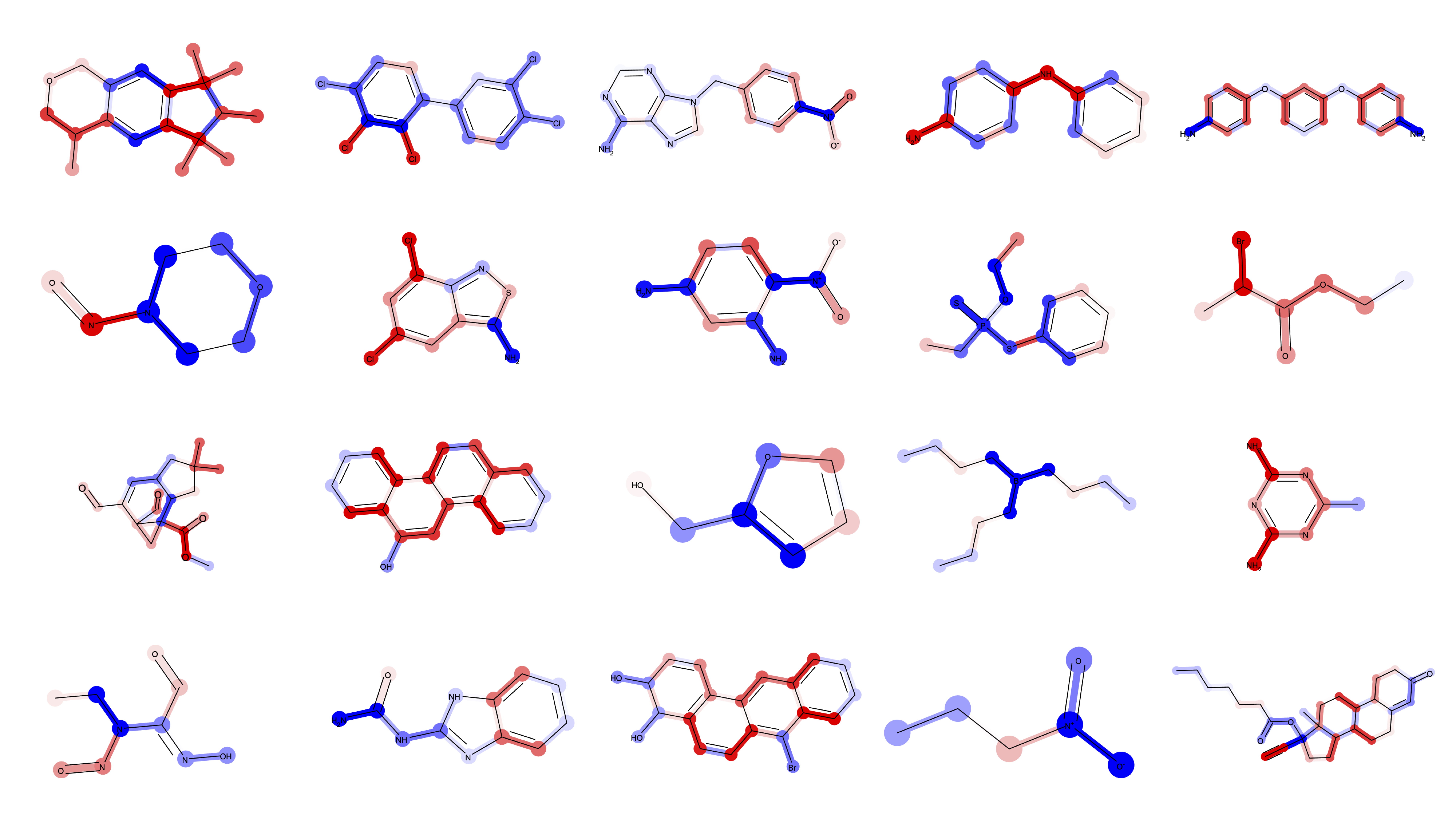
近年, 創薬・材料探索への深層学習の応用が着目されています. 特に, 分子構造を「グラフ」と見なしこれに深層学習を適用する「Graph Convolutional Network(GCN) 」は, 世界的な機械学習コンペティションであるKaggleの上位解法として用いられたことがあり, 注目を集めています.
本記事では, 深層学習により化合物の物性・薬理活性・毒性を予測し, その判断根拠を可視化するための解釈可能なGCNの教科書的な実装を一通り実演します. より具体的には, 実運用を想定して
- 変異原性 (遺伝子に対する毒性)予測の実施
- Pytorch Geometricを用いたGCNの実装
- Early Stopping の実装
- 予測根拠可視化のための, Captumを用いたIntegrated Gradientsの実装
を行いました. 完成すると上図のようにGCNを用いた変異原性の予測根拠の可視化が達成可能となります.
2. Graph Convolutional Network (GCN) とは?
Graph Convolutional Networkの理論的な話は本稿では触れません. Graph Convolutional Network による溶解度予測 (回帰)の記事や他の記事を参考にしてみてください.
3. 開発環境
- Miniconda
- RDkit 2019.09.3
- Pytorch Ver.1.8.1
- Pytorch geometric Ver.1.7.0 インストール方法
- Python Ver.3.8.10
- CPU
- Captum Ver.0.4.0
4. データセット
本記事で用いるデータセットは変異原性(遺伝子に対する毒性)データセットです. これは, Hansenらが2009年の論文で発表した変異原性予測のベンチマークとなるデータセットであり, ここには6512化合物のSMILESと, それぞれの化合物の変異原性のあり[1],なし[0]がラベルされ格納されています.
こちらのSupporting Informationから, 「smiles_cas_N6512.smi」をダウンロードしましょう.
5. 化合物の読み込み
from rdkit import Chem
import numpy as np
f = open('smiles_cas_N6512.smi', 'r', encoding='UTF-8')
rows = f.read().split('\n')
f.close()
mols, properties = [], []
for i, r in enumerate(rows):
mol = Chem.MolFromSmiles(r.split('\t')[0])
property = r.split('\t')[2]
if mol is None:
print(f'{i}: SMILES can not be converted to Mol object')
else:
mols.append(mol)
properties.append(property)
mols, properties = np.array(mols), np.array(properties, dtype=int)
以下のような出力が得られると思います.
1: SMILES can not be converted to Mol object
2: SMILES can not be converted to Mol object
3: SMILES can not be converted to Mol object
4: SMILES can not be converted to Mol object
5: SMILES can not be converted to Mol object
6: SMILES can not be converted to Mol object
6. データセットの分割
今回は実装の手法を学ぶ事を主軸としますので, 単純にデータセットをトレーニングセットとバリデーションセットのみに分割します.
from sklearn.model_selection import StratifiedKFold
skf = StratifiedKFold(n_splits=5, random_state=1640, shuffle=True)
train_idx, valid_idx = list(skf.split(mols, properties))[0]
分割したデータセットをそれぞれみてみましょう
from rdkit.Chem import Draw
Draw.MolsToGridImage(mols[train_idx].tolist()[:10], molsPerRow=5)
Draw.MolsToGridImage(mols[train_idx].tolist()[:10], molsPerRow=5)
7. Molオブジェクトをグラフ化する
Molオブジェクトのグラフ化には, 以前に作成した, DeepChemのグラフ構造化アルゴリズムをPytorch用に移植したmol2graph.pyを用います. 実装コードが非常に長いので, mol2graph.pyの全ての処理内容を把握している必要はありません.
mol2graph.pyによってグラフ化されたデータは, torch_geometric.data.data.Dataのデータ型を持っており, その中に原子同士の結合関係や各原子の特徴ベクトルが格納されています.
以下にmol2graph.pyのコードを貼り付けましたので, この実装コードをPythonファイルとして保存して外部ライブラリとして使用してください.
import numpy as np
from rdkit import Chem
import torch
from torch_geometric.data import Data
def one_of_k_encoding(x, allowable_set):
"""
Encodes elements of a provided set as integers.
Parameters
----------
x: object
Must be present in `allowable_set`.
allowable_set: list
List of allowable quantities.
Example
-------
>>> import deepchem as dc
>>> dc.feat.graph_features.one_of_k_encoding("a", ["a", "b", "c"])
[True, False, False]
Raises
------
`ValueError` if `x` is not in `allowable_set`.
"""
if x not in allowable_set:
raise Exception("input {0} not in allowable set{1}:".format(x, allowable_set))
return list(map(lambda s: x == s, allowable_set))
def one_of_k_encoding_unk(x, allowable_set):
"""
Maps inputs not in the allowable set to the last element.
Unlike `one_of_k_encoding`, if `x` is not in `allowable_set`, this method
pretends that `x` is the last element of `allowable_set`.
Parameters
----------
x: object
Must be present in `allowable_set`.
allowable_set: list
List of allowable quantities.
Examples
--------
>>> dc.feat.graph_features.one_of_k_encoding_unk("s", ["a", "b", "c"])
[False, False, True]
"""
if x not in allowable_set:
x = allowable_set[-1]
return list(map(lambda s: x == s, allowable_set))
def get_intervals(l):
"""For list of lists, gets the cumulative products of the lengths"""
intervals = len(l) * [0]
intervals[0] = 1# Initalize with 1
for k in range(1, len(l)):
intervals[k] = (len(l[k]) + 1) * intervals[k - 1]
return intervals
def safe_index(l, e):
"""Gets the index of e in l, providing an index of len(l) if not found"""
try:
return l.index(e)
except:
return len(l)
class GraphConvConstants(object):
"""This class defines a collection of constants which are useful for graph convolutions on molecules."""
possible_atom_list = [
'C', 'N', 'O', 'S', 'F', 'P', 'Cl', 'Mg', 'Na', 'Br', 'Fe', 'Ca', 'Cu','Mc', 'Pd', 'Pb', 'K', 'I', 'Al', 'Ni', 'Mn'
]
"""Allowed Numbers of Hydrogens"""
possible_numH_list = [0, 1, 2, 3, 4]
"""Allowed Valences for Atoms"""
possible_valence_list = [0, 1, 2, 3, 4, 5, 6]
"""Allowed Formal Charges for Atoms"""
possible_formal_charge_list = [-3, -2, -1, 0, 1, 2, 3]
"""This is a placeholder for documentation. These will be replaced with corresponding values of the rdkit HybridizationType"""
possible_hybridization_list = ["SP", "SP2", "SP3", "SP3D", "SP3D2"]
"""Allowed number of radical electrons."""
possible_number_radical_e_list = [0, 1, 2]
"""Allowed types of Chirality"""
possible_chirality_list = ['R', 'S']
"""The set of all values allowed."""
reference_lists = [
possible_atom_list, possible_numH_list, possible_valence_list,
possible_formal_charge_list, possible_number_radical_e_list,
possible_hybridization_list, possible_chirality_list
]
"""The number of different values that can be taken. See `get_intervals()`"""
intervals = get_intervals(reference_lists)
"""Possible stereochemistry. We use E-Z notation for stereochemistry
https://en.wikipedia.org/wiki/E%E2%80%93Z_notation"""
possible_bond_stereo = ["STEREONONE", "STEREOANY", "STEREOZ", "STEREOE"]
"""Number of different bond types not counting stereochemistry."""
bond_fdim_base = 6
def get_feature_list(atom):
possible_atom_list = GraphConvConstants.possible_atom_list
possible_numH_list = GraphConvConstants.possible_numH_list
possible_valence_list = GraphConvConstants.possible_valence_list
possible_formal_charge_list = GraphConvConstants.possible_formal_charge_list
possible_number_radical_e_list = GraphConvConstants.possible_number_radical_e_list
possible_hybridization_list = GraphConvConstants.possible_hybridization_list
# Replace the hybridization
from rdkit import Chem
#global possible_hybridization_list
possible_hybridization_list = [
Chem.rdchem.HybridizationType.SP, Chem.rdchem.HybridizationType.SP2,
Chem.rdchem.HybridizationType.SP3, Chem.rdchem.HybridizationType.SP3D,
Chem.rdchem.HybridizationType.SP3D2
]
features = 6 * [0]
features[0] = safe_index(possible_atom_list, atom.GetSymbol())
features[1] = safe_index(possible_numH_list, atom.GetTotalNumHs())
features[2] = safe_index(possible_valence_list, atom.GetImplicitValence())
features[3] = safe_index(possible_formal_charge_list, atom.GetFormalCharge())
features[4] = safe_index(possible_number_radical_e_list,
atom.GetNumRadicalElectrons())
features[5] = safe_index(possible_hybridization_list, atom.GetHybridization())
return features
def features_to_id(features, intervals):
"""Convert list of features into index using spacings provided in intervals"""
id = 0
for k in range(len(intervals-1)):
id += features[k] * intervals[k]
# Allow 0 index to correspond to null molecule 1
id = id + 1
return id
def id_to_features(id, intervals):
features = 6 * [0]
# Correct for null
id -= 1
for k in range(0, 6 - 1):
# print(6-k-1, id)
features[6 - k - 1] = id // intervals[6 - k - 1]
id -= features[6 - k - 1] * intervals[6 - k - 1]
# Correct for last one
features[0] = id
return features
def atom_to_id(atom):
"""Return a unique id corresponding to the atom type"""
features = get_feature_list(atom)
return features_to_id(features, intervals)
def atom_features(atom, bool_id_feat=False, explicit_H=False,use_chirality=False):
if bool_id_feat:
return np.array([atom_to_id(atom)])
else:
# concatnate all atom features
results_ = one_of_k_encoding_unk(
atom.GetSymbol(),
[
'C',
'N',
'O',
'S',
'F',
'Si',
'P',
'Cl',
'Br',
'Mg',
'Na',
'Ca',
'Fe',
'As',
'Al',
'I',
'B',
'V',
'K',
'Tl',
'Yb',
'Sb',
'Sn',
'Ag',
'Pd',
'Co',
'Se',
'Ti',
'Zn',
'H',
'Li',
'Ge',
'Cu',
'Au',
'Ni',
'Cd',
'In',
'Mn',
'Zr',
'Cr',
'Pt',
'Hg',
'Pb',
'Unknown'
]
)
results=results_ + \
one_of_k_encoding(
atom.GetDegree(),
[0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10]
) + \
one_of_k_encoding_unk(
atom.GetImplicitValence(),
[0, 1, 2, 3, 4, 5, 6]
) + \
[
atom.GetFormalCharge(), atom.GetNumRadicalElectrons()
] + \
one_of_k_encoding_unk(
atom.GetHybridization().name,
[
Chem.rdchem.HybridizationType.SP.name,
Chem.rdchem.HybridizationType.SP2.name,
Chem.rdchem.HybridizationType.SP3.name,
Chem.rdchem.HybridizationType.SP3D.name,
Chem.rdchem.HybridizationType.SP3D2.name
]
) + \
[atom.GetIsAromatic()]
# In case of explicit hydrogen(QM8, QM9), avoid calling `GetTotalNumHs`
if not explicit_H:
results = results + one_of_k_encoding_unk(
atom.GetTotalNumHs(),
[0, 1, 2, 3, 4]
)
if use_chirality:
try:
results = results + one_of_k_encoding_unk(
atom.GetProp('_CIPCode'),
['R', 'S']) + [atom.HasProp('_ChiralityPossible')]
except:
results = results + [False, False] + [atom.HasProp('_ChiralityPossible')]
return np.array(results)
def bond_features(bond, use_chirality=False):
from rdkit import Chem
bt = bond.GetBondType()
bond_feats = [
bt == Chem.rdchem.BondType.SINGLE, bt == Chem.rdchem.BondType.DOUBLE,
bt == Chem.rdchem.BondType.TRIPLE, bt == Chem.rdchem.BondType.AROMATIC,
bond.GetIsConjugated(),
bond.IsInRing()
]
if use_chirality:
bond_feats = bond_feats + one_of_k_encoding_unk(
str(bond.GetStereo()),
["STEREONONE", "STEREOANY", "STEREOZ", "STEREOE"]
)
return np.array(bond_feats)
def get_bond_pair(mol):
bonds = mol.GetBonds()
res = [[],[]]
for bond in bonds:
res[0] += [bond.GetBeginAtomIdx(), bond.GetEndAtomIdx()]
res[1] += [bond.GetEndAtomIdx(), bond.GetBeginAtomIdx()]
return res
def mol2vec(mol):
atoms = mol.GetAtoms()
bonds = mol.GetBonds()
node_f= [atom_features(atom) for atom in atoms]
edge_index = get_bond_pair(mol)
edge_attr = [bond_features(bond, use_chirality=False) for bond in bonds]
for bond in bonds:
edge_attr.append(bond_features(bond))
data = Data(
x=torch.tensor(node_f, dtype=torch.float), # shape [num_nodes, num_node_features] を持つ特徴行列
edge_index=torch.tensor(edge_index, dtype=torch.long), #shape [2, num_edges] と型 torch.long を持つ COO フォーマットによるグラフ連結度
edge_attr=torch.tensor(edge_attr,dtype=torch.float) # shape [num_edges, num_edge_features] によるエッジ特徴行列
)
return data
mol2graphのテスト
試しに1分子を読み込んでグラフ構造化データの様子を調べてみましょう
mol = mols[train_idx][5]
mol

import mol2graph
graph = mol2graph.mol2vec(mol)
print(type(graph))
print(graph.x.shape)
print(graph.x)
<class 'torch_geometric.data.data.Data'>
torch.Size([11, 75])
tensor([[ 1., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.,
0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.,
0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.,
0., 0., 0., 1., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.,
0., 0., 1., 0., 0., 0., 0., 0., 0., 0., 1., 0., 0., 0.,
0., 0., 0., 1., 0.],
...
...
...
このように, graphはtorch_geometric.data.data.Dataのクラスに属しており, graph.~~のように実行することで, グラフ化されたデータの中の様子を見ることが可能です.
graph.xは分子グラフの特徴行列に対応します. 形状は $n_{atoms} \times n_{features}$になっており, featureの次元数は75です. この75という数値は1原子からいくつの情報を取得しベクトル化 (今回の実装では原子の有する情報を one-hot encording により75次元に変換している) するかという処理に依存し, DeepChemから移植してきたmol2graph.pyのアルゴリズムが1原子から75次元の特徴ベクトルを取得するものであったというに過ぎません.
原子の隣接関係についても見ていきましょう.
print(graph.edge_index.shape)
print(graph.edge_index)
torch.Size([2, 22])
tensor([[ 0, 1, 1, 2, 2, 3, 3, 4, 4, 5, 5, 6, 6, 7, 5, 8, 8, 9,
8, 10, 6, 1],
[ 1, 0, 2, 1, 3, 2, 4, 3, 5, 4, 6, 5, 7, 6, 8, 5, 9, 8,
10, 8, 1, 6]])
ここでひとつ注意が必要なことは, graph.edge_indexによって得られる原子の隣接関係を表現するデータ構造についてです. 一般的に原子の隣接関係は隣接行列によって表現されますが, graph.edge_indexはその表現方法が異なり, $2 \times 2・n_{edges}$の形状をもちます. graph.edge_indexの1行目は$index_{begin}$, 2行目は$index_{end}$に対応し, 列数は辺の数($n_{edges}$)の2倍に対応します. つまり, 1行1列目と2行1列目はそれぞれ, 結合の始点となる原子のインデックスと結合の終点となる原子のインデックスに対応します.
8. データセットの作成
GCNに投げるためにMolオブジェクトをグラフ構造に変換します.
import torch
from torch_geometric.data import DataLoader
import mol2graph
if torch.cuda.is_available():
device = torch.device('cuda')
print('The code uses GPU!')
else:
device = torch.device('cpu')
print('The code uses CPU...')
X_train = [mol2graph.mol2vec(m) for m in mols[train_idx].tolist()]
y_train = properties[train_idx]
for i, data in enumerate(X_train):
data.y = torch.LongTensor([y_train[i]]).to(device)
X_valid = [mol2graph.mol2vec(m) for m in mols[valid_idx].tolist()]
y_valid = properties[valid_idx]
for i, data in enumerate(X_valid):
data.y = torch.LongTensor([y_valid[i]]).to(device)
train_loader = DataLoader(X_train, batch_size=128, shuffle=True, drop_last=True)
valid_loader = DataLoader(X_valid, batch_size=128, shuffle=True, drop_last=True)
今回の学習ではミニバッチ学習を採用しますので, DataLoaderを用いてデータをミニバッチに分割します. バッチサイズは特に理由はないですが128としました. DataLoaderによって生成された train_loaderはイテレータとして用いることが可能で, 以下のようにfor文で中身を確認できます. 確かに128サンプルずつバッチに詰められていることが分かります.
for i, d in enumerate(train_loader):
if i==3:
break
print(d.y)
print(d.y.shape)
tensor([1, 0, 1, 1, 1, 0, 1, 1, 1, 1, 0, 0, 1, 1, 1, 1, 1, 0, 1, 1, 0, 0, 1, 1,
0, 1, 1, 1, 0, 0, 0, 1, 0, 1, 1, 1, 1, 0, 0, 1, 1, 0, 1, 1, 0, 0, 1, 1,
0, 1, 0, 1, 1, 0, 0, 1, 0, 0, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 0, 0, 0, 0,
1, 0, 1, 1, 0, 1, 1, 0, 1, 1, 1, 1, 0, 0, 1, 1, 0, 1, 1, 0, 1, 0, 0, 1,
1, 1, 1, 1, 0, 1, 0, 1, 0, 0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0, 1, 1, 1, 0,
0, 0, 0, 0, 1, 0, 1, 0])
torch.Size([128])
tensor([1, 1, 1, 1, 0, 1, 0, 0, 1, 1, 1, 0, 1, 0, 0, 0, 1, 0, 0, 1, 0, 1, 1, 1,
1, 1, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 1, 0, 0, 0, 0, 1, 1, 1, 0, 1, 1, 1,
1, 1, 1, 0, 1, 0, 0, 1, 1, 1, 1, 1, 0, 1, 0, 0, 1, 1, 1, 1, 1, 1, 1, 1,
1, 0, 1, 0, 1, 1, 1, 1, 1, 1, 0, 1, 1, 0, 0, 1, 0, 1, 0, 0, 1, 1, 1, 0,
1, 1, 0, 1, 1, 1, 1, 1, 0, 1, 0, 1, 1, 0, 0, 1, 0, 1, 1, 1, 1, 1, 0, 0,
1, 1, 0, 1, 1, 1, 1, 0])
torch.Size([128])
tensor([1, 1, 1, 1, 1, 1, 1, 1, 1, 0, 1, 1, 0, 0, 1, 1, 0, 0, 0, 1, 0, 1, 1, 0,
1, 0, 1, 0, 0, 0, 0, 1, 0, 1, 1, 1, 1, 0, 0, 1, 0, 0, 0, 1, 1, 1, 1, 0,
0, 1, 0, 0, 1, 0, 1, 0, 0, 1, 0, 1, 1, 0, 0, 0, 1, 0, 1, 1, 1, 0, 1, 0,
0, 1, 0, 0, 1, 1, 1, 1, 0, 1, 0, 1, 0, 1, 1, 1, 0, 0, 1, 0, 0, 1, 0, 0,
0, 1, 0, 0, 0, 1, 1, 1, 0, 0, 0, 0, 1, 0, 0, 1, 1, 0, 1, 0, 1, 1, 0, 1,
1, 0, 0, 1, 1, 1, 1, 0])
torch.Size([128])
9. GCNクラスの設計
Pytorchの一般的な雛形に基づいてGCNモデルのクラス設計を行います.
from torch_geometric.nn import GCNConv
from torch_geometric.nn import global_add_pool
import torch.nn.functional as F
from torch.nn import ModuleList, Linear, BatchNorm1d
class MolecularGCN(torch.nn.Module):
def __init__(self, dim, n_conv_hidden, n_mlp_hidden, dropout):
super(MolecularGCN, self).__init__()
self.n_features = 75 # This is the mol2graph.py-specific value
self.n_conv_hidden = n_conv_hidden
self.n_mlp_hidden = n_mlp_hidden
self.dim = dim
self.dropout = dropout
self.graphconv1 = GCNConv(self.n_features, self.dim, cached=False)
self.bn1 = BatchNorm1d(self.dim)
self.graphconv_hidden = ModuleList(
[GCNConv(self.dim, self.dim, cached=False) for _ in range(self.n_conv_hidden)]
)
self.bn_conv = ModuleList(
[BatchNorm1d(self.dim) for _ in range(self.n_conv_hidden)]
)
self.mlp_hidden = ModuleList(
[Linear(self.dim, self.dim) for _ in range(self.n_mlp_hidden)]
)
self.bn_mlp = ModuleList(
[BatchNorm1d(self.dim) for _ in range(self.n_mlp_hidden)]
)
self.mlp_out = Linear(self.dim, 2)
def forward(self, x, edge_index, batch, edge_weight=None):
x = F.relu(self.graphconv1(x, edge_index, edge_weight))
x = self.bn1(x)
for graphconv, bn_conv in zip(self.graphconv_hidden, self.bn_conv):
x = graphconv(x, edge_index, edge_weight)
x = bn_conv(x)
x = global_add_pool(x, batch)
for fc_mlp, bn_mlp in zip(self.mlp_hidden, self.bn_mlp):
x = F.relu(fc_mlp(x))
x = bn_mlp(x)
x = F.dropout(x, p=self.dropout, training=self.training)
x = F.log_softmax(self.mlp_out(x), dim=-1)
return x
これでモデルクラスの設計は完了です. forward関数のレイヤー数などはモデルインスタンスを生成する際に簡単に変更できるように, ModuleListを使いあらかじめレイヤを複数個定義しておき, そこからfor文によりレイヤを付け足していく構造にしています.
また, 2クラス分類の場合は出力ノード数を1ノードにする場合もありますが, 今回は2ノードに設定しています.
10. モデルの学習と評価を行う関数の作成
モデルの学習と評価を行う関数を作っていきます.
from sklearn.metrics import roc_auc_score
def train(model, optimizer, loader):
model.train()
loss_all = 0
for data in loader:
optimizer.zero_grad()
data = data.to(device)
output = model.forward(data.x, data.edge_index, data.batch).squeeze(1)
loss = F.cross_entropy(output, data.y)
loss.backward()
loss_all += loss.item() * data.num_graphs
optimizer.step()
return loss_all / len(loader)
def eval(model, loader):
model.eval()
with torch.no_grad():
P, T, S = [], [], []
loss_all = 0
for data in loader:
data = data.to(device)
y_true = data.y.to(device).detach().numpy()
output = model.forward(data.x, data.edge_index, data.batch)
y_pred = output.detach().numpy()[:,1]
P.append(y_pred)
T.append(y_true)
S.append(roc_auc_score(y_true, y_pred))
loss = F.cross_entropy(output, data.y)
loss_all += loss.item() * data.num_graphs
return np.concatenate(T), np.concatenate(P), loss_all / len(loader), np.mean(S)
trainでは, モデルをmodel.train()により学習モードに設定し, ミニバッチ学習を行います. この際, マルチノード出力の場合, loss関数にcross entropy (F.cross_entropy()) を用いることに注意が必要です.
loss.backward()でバックプロパゲーションを実施し勾配を計算し, optimizer.step()で重みの更新を行います.
evalでは, モデルをmodel.eval()により評価モードに設定します. この時, torch.no_grad()により勾配の計算は行わないようにします. evalでは, 学習されたモデルを用いて損失の計算, 予測性能の計算などを行います. 学習において毎エポックごとにeval関数を実行し, エポックごとのモデルの予測性能を評価します.
11. Early stoppingクラスの実装
Pytorchを用いたDeep Learningの例でよく見かけるのは, ここからすぐに学習に移るケースです. しかし, Deep Learningを行う場合, Early stopping(学習の早期打ち切り)の機能を設定しなければ永遠と設定したepoch分計算し続けますし, ベストエポックの時の重みを保存できません. そのため過学習モデルが出来上がります.
そこで, Early stopping を実装することで過学習を抑制し, 学習を効率的に完了できるようにします. Pytorchは細かいアーキテクチャの設定などがしやすいため研究向きではありますが, kerasでは標準装備されている early bstoppingなども自前で用意しなければならないのが少し面倒ですね.
class EarlyStopping:
"""earlystoppingクラス"""
def __init__(self, patience=5, verbose=False, path='checkpoint_model.pth'):
"""引数:最小値の非更新数カウンタ、表示設定、モデル格納path"""
self.patience = patience #設定ストップカウンタ
self.verbose = verbose #表示の有無
self.counter = 0 #現在のカウンタ値
self.best_score = None #ベストスコア
self.early_stop = False #ストップフラグ
self.val_loss_min = np.Inf #前回のベストスコア記憶用
self.path = path #ベストモデル格納path
def __call__(self, val_loss, model):
"""
特殊(call)メソッド
実際に学習ループ内で最小lossを更新したか否かを計算させる部分
"""
score = -val_loss
if self.best_score is None: #1Epoch目の処理
self.best_score = score #1Epoch目はそのままベストスコアとして記録する
self.checkpoint(val_loss, model) #記録後にモデルを保存してスコア表示する
elif score < self.best_score: # ベストスコアを更新できなかった場合
self.counter += 1 #ストップカウンタを+1
if self.verbose: #表示を有効にした場合は経過を表示
print(f'EarlyStopping counter: {self.counter} out of {self.patience}') #現在のカウンタを表示する
if self.counter >= self.patience: #設定カウントを上回ったらストップフラグをTrueに変更
self.early_stop = True
else: #ベストスコアを更新した場合
self.best_score = score #ベストスコアを上書き
self.checkpoint(val_loss, model) #モデルを保存してスコア表示
self.counter = 0 #ストップカウンタリセット
def checkpoint(self, val_loss, model):
'''ベストスコア更新時に実行されるチェックポイント関数'''
if self.verbose: #表示を有効にした場合は、前回のベストスコアからどれだけ更新したか?を表示
print(f'Validation loss decreased ({self.val_loss_min:.6f} --> {val_loss:.6f}). Saving model ...')
torch.save(model.state_dict(), self.path) #ベストモデルを指定したpathに保存
self.val_loss_min = val_loss #その時のlossを記録する
EarlyStopping自体は機能すれば正直中身はなんでも良いので特に解説はしません. 使い方を実演しますので, そちらを理解すれば十分です.
12. モデルの学習
ようやく学習のための準備が完了しました.
# parameters
lr = 1e-4
n_epochs = 100
# model setting
model = MolecularGCN(dim = 64,n_conv_hidden = 4,n_mlp_hidden = 1,dropout = 0.1).to(device)
optimizer = torch.optim.Adam(model.parameters(), lr=lr)
earlystopping = EarlyStopping(patience=1, verbose=True)
# train
history = {
'loss_train':[],
'loss_valid':[],
'score_train':[],
'score_valid':[],
'y_true_train':[],
'y_true_valid':[],
'y_pred_train':[],
'y_pred_valid':[],
}
for epoch in range(1, n_epochs+1):
# training
train(model, optimizer, train_loader)
# performance evaluation
y_train, y_pred_train, loss_train, score_train = eval(model, train_loader)
y_valid, y_pred_valid, loss_valid, score_valid = eval(model, test_loader)
items = [
loss_train,
loss_valid,
score_train,
score_valid,
y_train,
y_valid,
y_pred_train,
y_pred_valid,
]
# save history
for i, item in enumerate(items):
keys = list(history.keys())
history[keys[i]].append(item)
print(f'Epoch: {epoch}/{n_epochs}, loss_train: {loss_train:.5},\
loss_valid: {loss_valid:.5}, AUC_train: {score_train:.5}, AUC_valid: {score_valid:.5}')
# early stopping detection
earlystopping(loss_valid, model)
if earlystopping.early_stop:
print("-"*100)
print("Early Stopping!")
print("-"*100)
break
Epoch: 1/100, loss_train: 85.095, loss_valid: 84.239, AUC_train: 0.69868, AUC_valid: 0.69917
Validation loss decreased (inf --> 84.239291). Saving model ...
Epoch: 2/100, loss_train: 77.081, loss_valid: 76.886, AUC_train: 0.74935, AUC_valid: 0.74176
Validation loss decreased (84.239291 --> 76.886169). Saving model ...
Epoch: 3/100, loss_train: 74.06, loss_valid: 74.432, AUC_train: 0.77052, AUC_valid: 0.75815
Validation loss decreased (76.886169 --> 74.431856). Saving model ...
Epoch: 4/100, loss_train: 72.155, loss_valid: 73.292, AUC_train: 0.79146, AUC_valid: 0.77076
Validation loss decreased (74.431856 --> 73.291620). Saving model ...
Epoch: 5/100, loss_train: 70.754, loss_valid: 71.884, AUC_train: 0.8009, AUC_valid: 0.7822
Validation loss decreased (73.291620 --> 71.884274). Saving model ...
Epoch: 6/100, loss_train: 69.483, loss_valid: 70.793, AUC_train: 0.81026, AUC_valid: 0.79152
Validation loss decreased (71.884274 --> 70.793071). Saving model ...
Epoch: 7/100, loss_train: 68.396, loss_valid: 70.498, AUC_train: 0.81725, AUC_valid: 0.79156
Validation loss decreased (70.793071 --> 70.497758). Saving model ...
Epoch: 8/100, loss_train: 67.533, loss_valid: 69.829, AUC_train: 0.8242, AUC_valid: 0.79937
Validation loss decreased (70.497758 --> 69.829383). Saving model ...
Epoch: 9/100, loss_train: 66.523, loss_valid: 69.188, AUC_train: 0.82781, AUC_valid: 0.80461
Validation loss decreased (69.829383 --> 69.187917). Saving model ...
Epoch: 10/100, loss_train: 65.658, loss_valid: 68.787, AUC_train: 0.83369, AUC_valid: 0.80712
Validation loss decreased (69.187917 --> 68.786839). Saving model ...
Epoch: 11/100, loss_train: 65.014, loss_valid: 67.933, AUC_train: 0.838, AUC_valid: 0.81469
Validation loss decreased (68.786839 --> 67.933392). Saving model ...
Epoch: 12/100, loss_train: 64.63, loss_valid: 67.566, AUC_train: 0.83844, AUC_valid: 0.81522
Validation loss decreased (67.933392 --> 67.566029). Saving model ...
Epoch: 13/100, loss_train: 63.536, loss_valid: 67.109, AUC_train: 0.84579, AUC_valid: 0.81903
Validation loss decreased (67.566029 --> 67.108937). Saving model ...
Epoch: 14/100, loss_train: 62.887, loss_valid: 66.278, AUC_train: 0.84787, AUC_valid: 0.82399
Validation loss decreased (67.108937 --> 66.277645). Saving model ...
Epoch: 15/100, loss_train: 62.37, loss_valid: 66.01, AUC_train: 0.85119, AUC_valid: 0.82655
Validation loss decreased (66.277645 --> 66.009877). Saving model ...
Epoch: 16/100, loss_train: 62.188, loss_valid: 65.269, AUC_train: 0.85516, AUC_valid: 0.8323
Validation loss decreased (66.009877 --> 65.268563). Saving model ...
Epoch: 17/100, loss_train: 61.419, loss_valid: 65.126, AUC_train: 0.85748, AUC_valid: 0.83273
Validation loss decreased (65.268563 --> 65.125996). Saving model ...
Epoch: 18/100, loss_train: 61.18, loss_valid: 65.692, AUC_train: 0.8574, AUC_valid: 0.82943
EarlyStopping counter: 1 out of 1
----------------------------------------------------------------------------------------------------
Early Stopping!
----------------------------------------------------------------------------------------------------
学習過程に関しては雛形通りにfor文でエポック数回すだけです. この中でトレーニングセットとバリデーションセットに対するlossと予測性能をeval関数を用いてモニターします.
ポイントはearly stoppingが導入されている点です.earlystopping = EarlyStopping(patience=1, verbose=True)の部分で, for文に入る前にあらかじめearlystoppingをインスタンス化し, この際にpatienceを設定します.
patienceを超えてlossが最小値をカウントしないエポックが続いた場合, 学習をそこで打ち切ります. early stoppingのモニターはfor文内のearlystopping(loss_valid, model)で行っており, 最小lossをカウントするごとにモデルの重みを保存しています. デフォルトでは"checkpoint_model.pth"という名前で保存されますが, 変更することも可能です.
early stopping の判定結果はearlystopping.early_stopによりbool値で返ってきます. これがTrueの時にfor文を打ち切っています.
今回は簡易化のためpatience=1としましたが, 実用ではエポック数を無限にしてpatienceは学習不足を回避するために学習率の逆数程度(1000とかを著者はよく用います)に設定すると良いです. patienceは高過ぎても学習が終わりませんので時間とマシンスペックと相談して決めると良いと思います.
13. ベストエポックの重みのロードとモデルの評価
学習が終わったので, ベストエポックの重みをロードして学習過程の可視化とモデル評価を行います.
# draw learing curve
import matplotlib.pyplot as plt
fig = plt.figure(figsize=(8,6))
ax1 = fig.add_subplot(111)
ax2 = ax1.twinx()
epochs_ = np.arange(1,len(history['loss_train'])+1)
ax1.plot(epochs_, history['loss_train'], label="loss_train", c='blue')
ax2.plot(epochs_, history['loss_valid'], label=r"loss_valid", c='green')
ax1.set_xlabel('epochs')
ax1.set_ylabel(r'loss')
ax1.grid(True)
h1, l1 = ax1.get_legend_handles_labels()
h2, l2 = ax2.get_legend_handles_labels()
ax2.legend(h1+h2, l1+l2, loc='lower right')
plt.show()
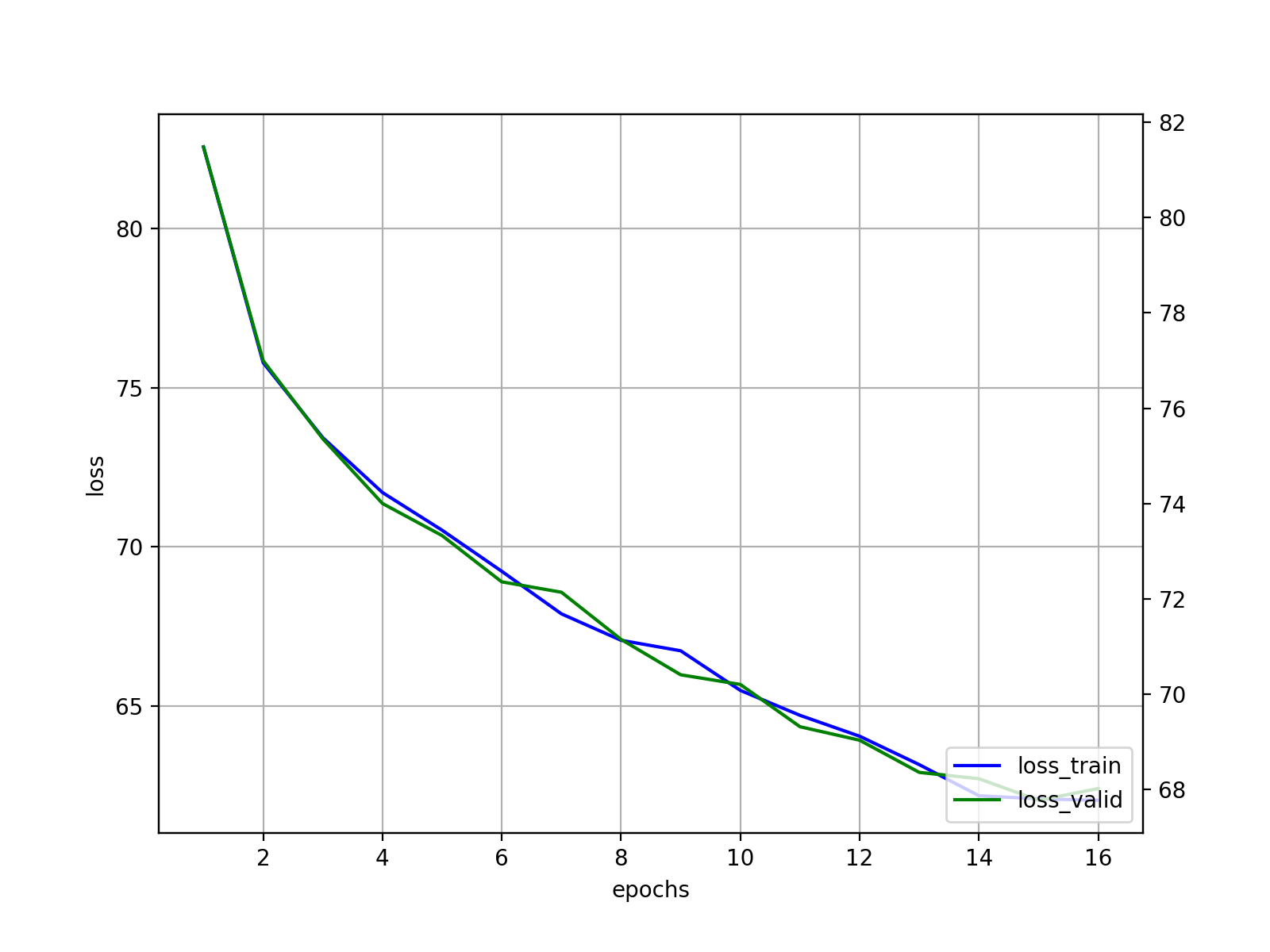
training setのlossとvalidation setのlossがパラレルに減少しており, きちんと学習できています. 確かにearly stoppingのおかげで過学習はしていないようですね.
from sklearn.metrics import roc_curve
# load best weight
model.load_state_dict(torch.load('checkpoint_model.pth'))
# prediction with best weight
y_train, y_pred_train, loss_train, score_train = eval(model, train_loader)
y_valid, y_pred_valid, loss_valid, score_valid = eval(model, test_loader)
# evaluation
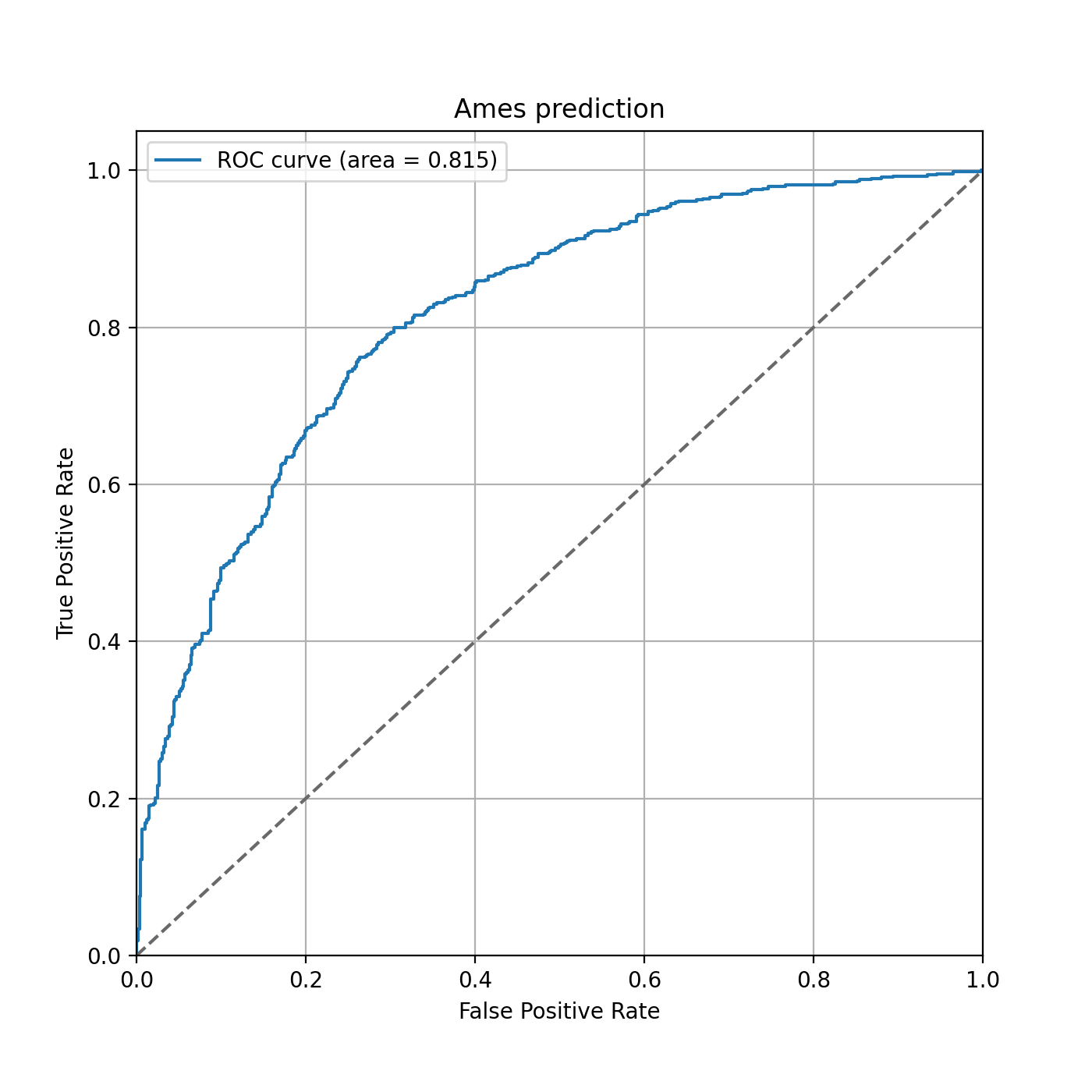
fpr, tpr, thresholds = roc_curve(y_valid, y_pred_valid)
auc = roc_auc_score(y_valid, y_pred_valid)
# drawing
plt.figure(figsize=(7,7))
plt.plot(fpr, tpr, label='ROC curve (area = %0.3f)' % auc)
plt.plot([0, 1], [0, 1], color='dimgrey', linestyle='--')
plt.xlim([0.0, 1.0])
plt.ylim([0.0, 1.05])
plt.xlabel('False Positive Rate')
plt.ylabel('True Positive Rate')
plt.title('Ames prediction')
plt.legend()
plt.grid(True)
plt.show()
14. 予測根拠の可視化
GCNの予測根拠の可視化を行います. 実装にはCaptumというライブラリを用いて行っていきます. 今回は, Deep learningによる予測根拠を可視化する手法として一般的に用いられているIntegrated Gradientsを用います.
14.1. Integrated Gradients
参照元 Axiomatic Attribution for Deep Networks
14.1.1. モチベーション
線形モデルの場合, モデルを解釈するためにモデル係数を検査します. そのため, ディープネットワークモデルの係数(勾配)を考えることは線形モデルにおけるモデル解釈の発想をDeep learningモデルに転換する自然なアナロジーとなります. そのため, モデルの勾配を取得することはDeep learningモデルの予測根拠解釈の合理的な出発点となります.
ただ, 単純に勾配を計算した場合, 非線形変換を用いているDeep Learningでは, 入力$x$の値によっては勾配が0になるケースが存在します. 例えば, $f(x)=1− ReLU(1−x)$ のケースを考えてみると, 入力$x$が1以上の時に勾配が0になります. この時, 入力$x$の寄与は0となってしまいます.
このような事態を回避するために, Integrated Gradientsという手法が導入されました. Integrated Gradientsは既存のDeep Learningモデルに変更を加えることなく実装できるため, 手軽であるという利点も有します.
14.1.2. 理論概要
Integrated Gradientsは以下の式で定式化されます.
$$Integrated \ Grads_i = (x_i - x^{\prime}_i)\times \int^1_0 \frac{{\partial}(F(x^{\prime}+{\alpha}(x-x^{\prime})))}{{\partial}x_i} d{\alpha}$$ ここで, $x^{\prime}$はベースラインの入力, $x$は所望のサンプルの入力, $x_i$は入力$x$の$i$番目の次元の値, ${\alpha}$は摂動係数を指します. イメージとしては, ベースライン入力とサンプル入力を重ねたもの$x^{\prime}+{\alpha}(x-x^{\prime})$をモデルに投入し$i$番目のfeatureについて微分した際の局所勾配を算出し, この作業を${\alpha}=0$から${\alpha}=1$ の範囲で変化させ(サンプルの入力条件を変化させ)全て足し合わせる. これを, ベースライン入力とサンプル入力の差を示す $(x_i - x^{\prime}_i)$に乗じることで, $x_i$の貢献度が計算されるということです.
GCNの場合, ベースラインを0と取ると上記の式は非常にシンプルなものとなります.
$$Integrated \ Grads_i = x_i\times \int^1_0 \frac{{\partial}F({\alpha}x)}{{\partial}x_i} d{\alpha}$$
実際の計算はこれを離散的に近似して計算しています.
14.1.3. 実装
まず必要な関数群を定義していきます.
from captum.attr import IntegratedGradients
from collections import defaultdict
def integrated_gradients_edge_mask(model, data, target):
def model_forward(edge_mask, data):
batch = torch.zeros(data.x.shape[0], dtype=int).to(device)
out = model(data.x, data.edge_index, batch, edge_mask)
return out
# integrated gradients
input_mask = torch.ones(data.edge_index.shape[1]).requires_grad_(True).to(device)
ig = IntegratedGradients(model_forward)
mask = ig.attribute(
input_mask,
target=target,
additional_forward_args=(data,),
internal_batch_size=data.edge_index.shape[1]
)
edge_mask = mask.cpu().detach().numpy()
# aggregate edge directions
edge_mask_dict = defaultdict(float)
for val, u, v in list(zip(edge_mask, *data.edge_index)):
u, v = u.item(), v.item()
if u > v:
u, v = v, u
edge_mask_dict[(u, v)] += val
edge_mask_dict_max = max(np.abs([_ for _ in edge_mask_dict.values()]))
if edge_mask_dict_max > 0:
edge_mask_dict = {key: value/edge_mask_dict_max for key, value in edge_mask_dict.items()}
return edge_mask_dict
def edge_mask_to_atom_mask(edge_mask_dict):
# get atom indexes
atom_indexes = []
for key in edge_mask_dict.keys():
i, j = key[0], key[1]
atom_indexes.append(i)
atom_indexes.append(j)
# integrate edge masks into atom
atom_mask_dict = {}
for i in range(max(atom_indexes)+1):
i_values = []
for key, value in edge_mask_dict.items():
if i in key:
i_values.append(value)
atom_mask_dict[i] = i_values[np.argmax(np.abs(i_values))]
return atom_mask_dict
def bond_pairs_to_id (edge_index):
bond_pairs = {}
for i in range(edge_index.shape[1]):
if i%2==0:
bond_pair = sorted([edge_index[0][i], edge_index[1][i]])
bond_pairs[int(i/2)] = (bond_pair[0], bond_pair[1])
return bond_pairs
def calor_cmap(x):
"""Red to Blue color map
x: list
"""
cmaps = []
for v in x:
if v > 0:
# Red cmap for positive value
cmap = (1.0, 1.0 - v, 1.0 - v)
else:
# Blue cmap for negative value
v *= -1
cmap = (1.0 - v, 1.0 - v, 1.0)
cmaps.append(cmap)
return cmaps
こちらもプログラムが長いですが, 殆どはカラーマップを作成するためにIntegrated Gradientsの結果をどのようにまとめるかという処理に該当します.
Integrated Gradientsの処理の本体はintegrated_gradients_edge_mask関数の初めの数行です.
- まずintegrated Gradientsに投入する用の$F$に該当する関数を作ります.今回は, GCNによる予測の際に
edge_weightを全て1にするために, モデルをラップしたforwardを定義しています.input_maskはedge_weghtに1を使用するように設定しているだけです. - 次に
IntegratedGradientsをインスタンス化する際に$F$に該当する関数を引数に入れます. -
ig.attribute()でIntegrated Gradientsの結果が返ります. 第一引数には$F$の第一引数が入り,additional_forward_argsで$F$が他にとりうる引数を設定することができます. -
targetにはそのサンプルの予測クラスが入ります. 今回は正解クラスがわかっているのでそれを直接入力していますが, ここにGCNによる予測クラスを入力すれば, その化合物をそのクラスに分類した根拠がわかります. - デフォルトではベースラインは0になります.
これによって, エッジのIG(Integrated Gradients)を示す結果が返ってくるため、どの原子が関与しているか? という情報をカラーマップにするためにはその原子に集約しているエッジの貢献度から何らかの処理(平均化)などを噛ませる必要があります.今回は便宜的に, 原子に集約している全てのエッジの中で最もIGの絶対値が大きいエッジのIGをその原子のカラーとして採用しています.
実際に可視化してみましょう.
# データを適当に選びます.
data = X_valid[11]
mol = mols[test_idx][11]
# Integrated Gradientsの結果を, カラーマップで示すために辞書型にします.
edge_mask_dict = integrated_gradients_edge_mask(model, data, data.y)
edge_pairs_dict = bond_pairs_to_id(data.edge_index.detach().numpy())
edge_ig_dict = {key: edge_mask_dict[value] for key, value in edge_pairs_dict.items() }
edge_igcolor_dict = {key: calor_cmap(list(edge_ig_dict.values()))[i] for i, key in enumerate(edge_ig_dict.keys())}
atom_mask_dict = edge_mask_to_atom_mask(edge_mask_dict)
atom_igcolor_dict = {key: calor_cmap(list(atom_mask_dict.values()))[i] for i, key in enumerate(atom_mask_dict.keys())}
from rdkit.Chem.Draw import rdMolDraw2D
from rdkit.Chem.Draw.MolDrawing import DrawingOptions
drawer = rdMolDraw2D.MolDraw2DSVG(600, 300)
drawer.drawOptions().padding = .0
drawer.SetLineWidth(2)
drawer.SetFontSize(.6)
drawer.drawOptions().updateAtomPalette({k: (0, 0, 0) for k in DrawingOptions.elemDict.keys()})
drawer.DrawMolecule(
rdMolDraw2D.PrepareMolForDrawing(mol),
highlightBonds = list(edge_igcolor_dict.keys()),
highlightBondColors=edge_igcolor_dict,
highlightAtoms=[k for k in atom_igcolor_dict .keys()],
highlightAtomColors=atom_igcolor_dict ,
highlightAtomRadii={i: 0.3 for i in range(len(atom_igcolor_dict))}
)
drawer.FinishDrawing()
svg = drawer.GetDrawingText().replace('svg:', '')
SVG(svg)
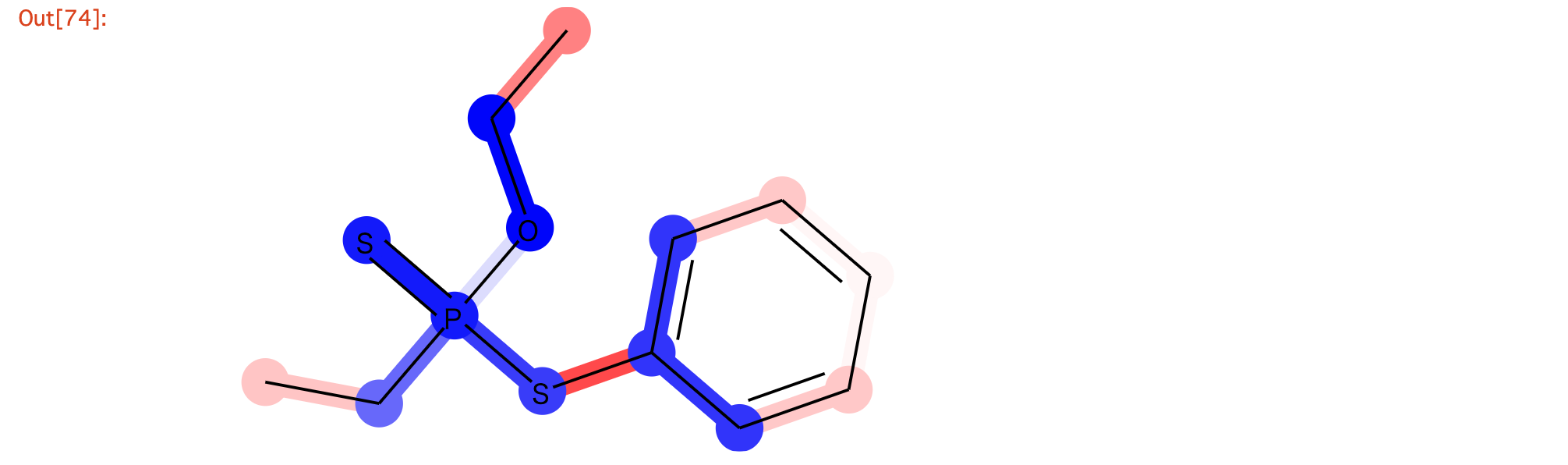
こんな感じで予測根拠を可視化できました.
15. 終わりに
変異原性予測を例にGCNによる予測と予測根拠の可視化を達成しました. 特に予測根拠の可視化の部分では, IGを計算した後の処理において, 視認性を向上させるためにさらなる変更の余地があると思いますので, いくつか試してみてください.