GEMMIとは
GEMMIは、主にマクロ分子結晶学(MX)で使用するために開発されたライブラリおよびツール群です。以下のような多彩なデータを扱うことができます:
- マクロ分子モデル(PDB、PDBx/mmCIF、mmJSON形式)
- 精密化の拘束条件(CIFファイル)
- 反射データ(MTZおよびmmCIF形式)
- 3Dグリッドデータ(電子密度マップ、マスク、MRC/CCP4形式)
- 結晶学的対称性
また、GEMMIは次のような特徴を持っています:
- 最速のオープンソースCIFパーサーを提供し、CIFファイルを使用する分子構造科学全般で有用
- 構造バイオインフォマティクスにも適しており、対称性を考慮したタンパク質モデルの解析が可能
- 移植性が高い:Linux、Windows、MacOSで動作し、WebAssembly経由でブラウザ内でも動作
- 多言語対応:C++14で記述され、Python(3.8以上)や一部C/Fortranとのインターフェースを提供
GEMMIはGlobal Phasing Ltd.とCCP4の共同プロジェクトであり、名前は「Gemmi Pass」に由来します。また、GEneral MacroMolecular I/o(一般的なマクロ分子入出力)の略とも解釈できます。
公式リポジトリ:GEMMI GitHub
Pythonでの利用について
GEMMIは主にC++で書かれていますが、Pythonバインディングが用意されており、Pythonからも簡単に利用できます。
この記事では、Python APIを使用した例を中心に説明します。
さらに、GEMMIはコマンドラインツールとしても利用可能です。詳細は公式ドキュメントをご参照ください。
構文ベースと構造モデルベース
GEMMIには、mmCIFファイルを解析するための二つのモードがあります。それぞれ用途が異なるため、目的に応じて使い分けることができます。
1. 構文ベース (Syntactic)
CIFファイルを構文レベルで読み取り・編集するモードです。このモードを使用すると、CIFファイル内のデータを直接操作できます。
具体的には、以下のような用途で便利です:
- CIFファイルから情報を取得する
- 特定の項目を追加・変更する
- 生データをそのまま解析したい場合
このモードについては、別の記事も書いていますのでそちらをご覧ください。
参考
https://gemmi.readthedocs.io/en/latest/cif.html#cif-parser
2. 構造モデルベース (Structure)
CIFファイル(PDBやmmJSONも対応)を、GEMMI独自のデータ構造に変換して解析するモードです。
このデータ構造は、以下の階層を持つ「SMRCAモデル」に基づいています:
- Structure → Model → Chain → Residue → Atom
このモードでは、構造情報を抽象化して扱うことができ、以下のような用途に適しています:
- タンパク質や他の分子モデルを解析する
- BioPythonの
Bio.PDBモジュールのように、構造情報を階層的に操作する
参考
https://gemmi.readthedocs.io/en/latest/mol.html#molecular-models
本記事では、後者の構造モデルベースの解析方法の基礎的な部分を解説していきます!!
ファイルの読み込み・書き込み
このセクションでは、GEMMIを使用して構造データを読み書きする方法を簡単に解説します。GEMMIの強力なパーサーを活用すれば、大規模なファイルでも高速に処理できます。
構造情報のダウンロード
GEMMIには、Bio.PDBのretrieve_pdb_file()のように直接ファイルをダウンロードする機能はありません。そのため、PDBファイルなどの必要なデータは、手動でダウンロードして準備する必要があります。
ただし、PDBデータをローカルに保存している場合、GEMMIのgemmi.expand_if_pdb_code()を使うと、特定のPDB IDに対応するローカルパスを自動生成できます。
特に、PDBデータをローカルにミラーリングしている環境では便利です。
import gemmi
import os
# 環境変数 $PDB_DIR を設定
os.environ['PDB_DIR'] = 'test'
# PDB ID '1alk' のパスを生成
path = gemmi.expand_if_pdb_code('1alk', 'M')
print(path)
test/structures/divided/mmCIF/al/1alk.cif.gz
このパスは、PDBデータが以下のようにローカルに保存されている場合に対応します:
$PDB_DIR/structures/divided/mmCIF/<先頭2文字>/<PDB ID>.cif.gz
構造データの読み込み
このセクションでは、GEMMIを使用した構造解析におけるファイルの読み書きについて簡単に説明します。
GEMMIの高速性
GEMMIのパーサーは非常に高速です。以下のコードで、Bio.PDBのFastMMCIFParser.get_structure()と比較してみましょう。
実行時間は実行環境によって異なります。
import time
import os
from Bio.PDB import FastMMCIFParser
from gemmi import read_structure
# Bio.Pythonのパーサー
parser = FastMMCIFParser(QUIET=True)
# ファイルサイズの確認
print('1alk.cif', 'file size:', f"{os.path.getsize('1alk.cif'):,}", 'bytes')
# Bio.Pythonの読み込み
start = time.time()
st = parser.get_structure('1alk', '1alk.cif')
print('BioPython:', f'{time.time() - start:.3f} sec')
# GEMMIの読み込み
start = time.time()
st = read_structure('1alk.cif')
print('GEMMI:', f'{time.time() - start:.3f} sec')
1alk.cif file size: 697,111 bytes
BioPython: 0.125 sec
GEMMI: 0.005 sec
GEMMIの高速性が際立っていますね.
大規模ファイルでの比較
1ALK は
- Total Structure Weight: 94.69 kDa
- Atom Count: 6,630
- Modelled Residue Count: 898
- Deposited Residue Count: 898
- Unique protein chains: 1
程度の大きさのエントリーなので、もっと大きな構造で比較してみましょう。
より大きな構造データとして、8GLV を使った比較を行います。このエントリーは以下のような非常に大きな構造データです:
- Total Structure Weight: 63,486.35 kDa
- Atom Count: 3,968,189
- Modelled Residue Count: 503,221
- Deposited Residue Count: 566,853
- Unique protein chains: 127
# 8GLVの読み込み
print('1alk.cif', 'file size:', f"{os.path.getsize('1alk.cif'):,}", 'bytes')
# BioPython
start = time.time()
st = parser.get_structure('8glv', '8glv.cif')
print('BioPython:', f'{time.time() - start:.3f} sec')
# GEMMI
start = time.time()
st = read_structure('8glv.cif')
print('GEMMI:', f'{time.time() - start:.3f} sec')
8glv.cif file size: 453,410,659 bytes
BioPython: 21.574 sec
GEMMI: 2.971 sec
ファイルサイズが大きいほど、GEMMIのパフォーマンスが際立ちます。
.cif.gz形式での読み込み
GEMMIでは、.cif.gz形式の圧縮ファイルもそのまま読み込めます。
import os
from gemmi import read_structure
# st = parser.get_structure('8glv', '8glv.cif.gz')
# BioPythonの場合gzは読み込めない
print('8glv.cif', 'file size:', f"{os.path.getsize('8glv.cif'):,}", 'bytes')
start = time.time()
st = read_structure('8glv.cif.gz')
print('time:', f'{time.time() - start:.3f} sec')
8glv.cif.gz file size: 87,739,484 bytes
time: 3.350 sec
その他のフォーマット
.cif.gzの他にも、GEMMIは様々なフォーマットに対応しています。
以下はその一覧です:
| フォーマット名 | 説明 |
|---|---|
CoorFormat.Unknown |
ファイル拡張子からフォーマットを推測(デフォルト) |
CoorFormat.Detect |
ファイル内容からフォーマットを推測(内容がCIFでもJSONでもない場合はPDBと見なされる) |
CoorFormat.Pdb |
PDB形式 |
CoorFormat.Mmcif |
mmCIF形式 |
CoorFormat.Mmjson |
mmJSON形式 |
CoorFormat.ChemComp |
リガンドやモノマーライブラリのCIFファイル。mmCIFと同じ拡張子を持つため、DetectまたはChemCompの指定が必要。 |
# PDB
>>> read_structure('1alk.pdb')
<gemmi.Structure 1alk with 1 model(s)>
>>> gemmi.read_pdb('1alk.pdb') # read_pdbもある
<gemmi.Structure 1alk with 1 model(s)>
with open('1alk.pdb', 'r') as f:
pdb_str = f.read()
>>> gemmi.read_pdb_string(pdb_str) # read_pdb_stringは文字列のPDBフォーマットを読み込める
# mmJSON
>>> read_structure('1alk.json')
<gemmi.Structure 1alk with 1 model(s)>
# ChemComp
>>> read_structure('ATP.cif') # 指定しない場合
<gemmi.Structure ATP with 0 model(s)> # モデルなし
>>> from gemmi import CoorFormat
>>> read_structure('ATP.cif', format=CoorFormat.ChemComp) # formatを指定
<gemmi.Structure ATP with 2 model(s)> # idealとmodelが読み込まれる
# DETECT
>>> import shutil
>>> shutil.copy('1alk.cif', '1alk.txt') # .cifを.txtに変換
>>> read_structure('1alk.txt') # CoorFormat.Unknownの場合
RuntimeError: Unknown format of 1alk.txt. # エラー
>>> read_structure('1alk.txt', format=CoorFormat.Detect) # CoorFormat.Detectを指定
<gemmi.Structure 1ALK with 1 model(s)> # ファイル内容から読み込んでくれる
詳細は公式ドキュメント(stable)をご覧ください。
構造の書き込み
構造の書き込みについても簡単に確認しておきましょう。
基本的に、新しいmmCIFファイルに構造を書き込むためには、gemmi.cif.Document (もしくはgemmi.cif.Block)に変換する必要があります。
1ALK からChain Bを削除して1alk_remove_B.cifを作ってみましょう
import gemmi
st = gemmi.read_structure('1alk.cif.gz')
print('削除前', list(st[0]))
del st[0]['B'] # Chain Bを削除
print('削除後', list(st[0]))
st.name = '1alk_remove_B' # Structureの名前を変更
doc = st.make_mmcif_document() # Structure -> Document
# block = st.make_mmcif_block() # Blockの場合
print('新しいDocument:', doc)
doc.write_file('1alk_remove_B.cif')
# block.write_file('1alk_remove_B.cif') # Blockの場合
削除前 [<gemmi.Chain A with 456 res>, <gemmi.Chain B with 456 res>]
削除後 [<gemmi.Chain A with 456 res>]
新しいDocument: <gemmi.cif.Document with 1 blocks (1alk_remove_B)>
ChimeraXなどのViewerで見てみると
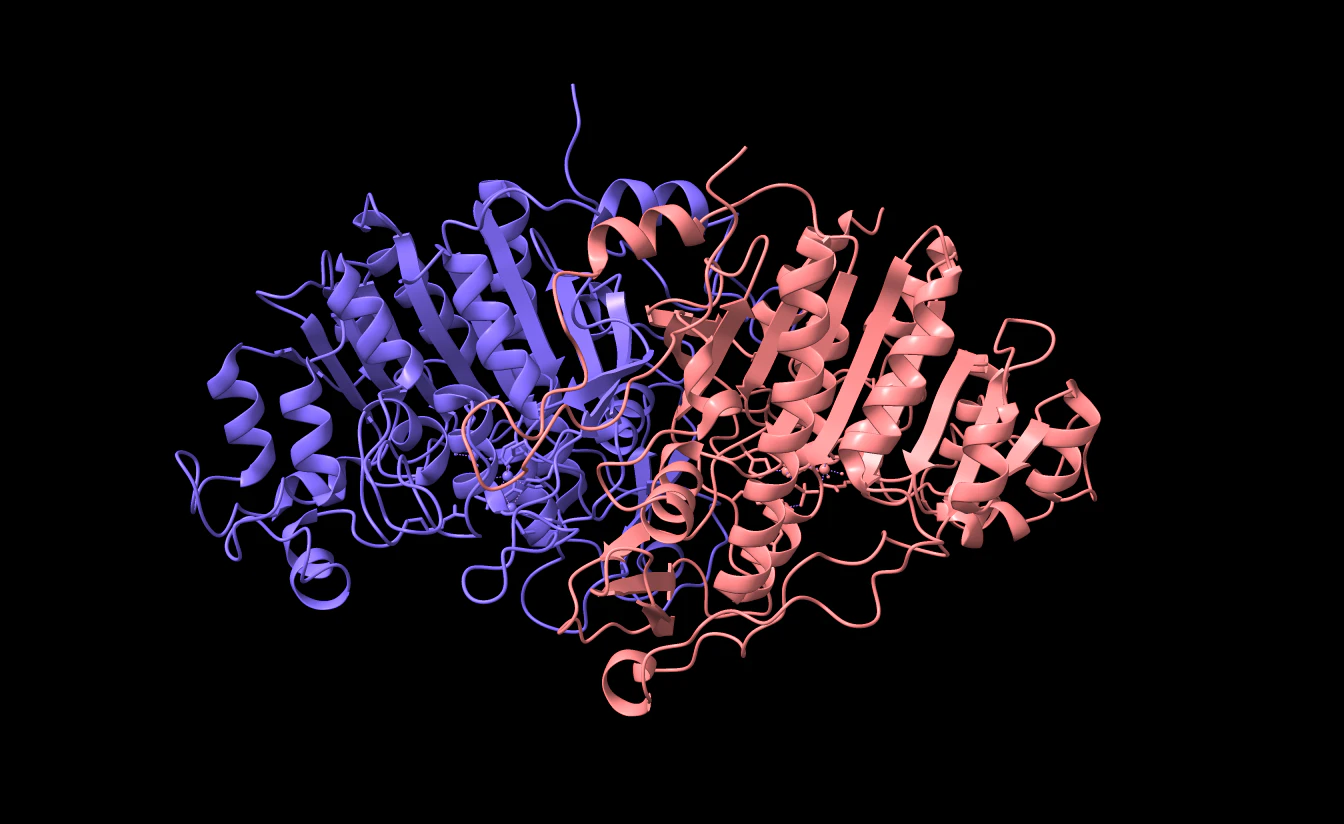
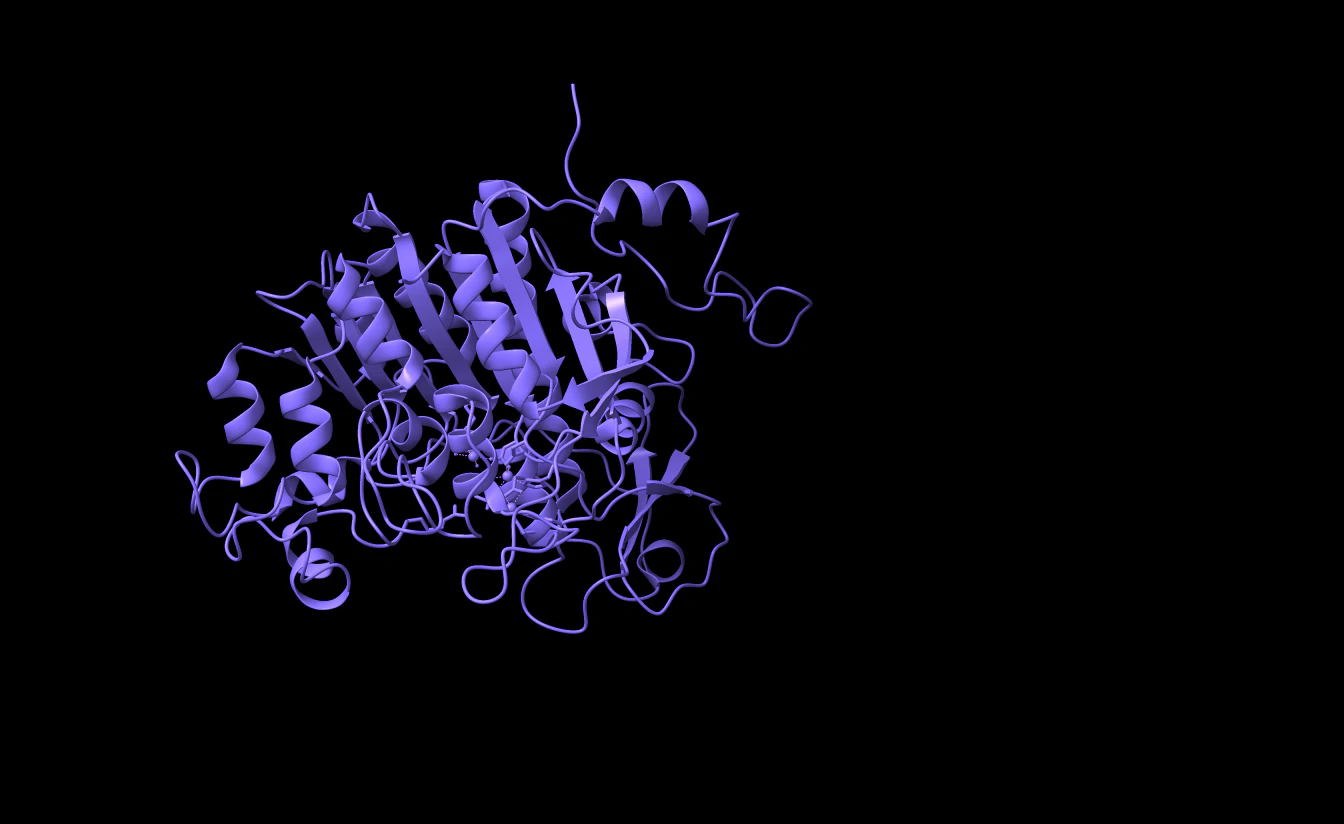
( <- 削除前 | 削除後 -> )
ChainB(ピンク) が削除されていますね
他にもgemmi.MmcifOutputGroupsを指定することで特定のカテゴリだけを書き込むこんだりすることができます。
import gemmi
st = gemmi.read_structure('1alk.cif.gz')
del st[0]['B']
st.name = '1alk_A_atoms'
groups = gemmi.MmcifOutputGroups(False) # Falseにすることで全てのカテゴリを無効化
groups.atoms = True # _atom_site を有効化
groups.block_name = True # _atom_site のブロック名を有効化
doc = st.make_mmcif_document(groups) # groups引数にMmcifOutputGroups
doc.write_file('1alk_A_atoms.cif')
print(doc.as_string()) # as_stringで書き込む内容を文字列で取得できます
data_1alk_A_atoms
loop_
_atom_site.id
_atom_site.type_symbol
_atom_site.label_atom_id
_atom_site.label_alt_id
_atom_site.label_comp_id
_atom_site.label_asym_id
_atom_site.label_entity_id
_atom_site.label_seq_id
_atom_site.pdbx_PDB_ins_code
_atom_site.Cartn_x
_atom_site.Cartn_y
_atom_site.Cartn_z
_atom_site.occupancy
_atom_site.B_iso_or_equiv
_atom_site.pdbx_formal_charge
_atom_site.auth_seq_id
_atom_site.auth_asym_id
_atom_site.pdbx_PDB_model_num
1 N N . THR A 1 1 ? 67.51 52.432 1.1 1 53.47 ? 1 A 1
2 C CA . THR A 1 1 ? 66.663 52.938 2.184 1 53.8 ? 1 A 1
3 C C . THR A 1 1 ? 66.66 51.955 3.361 1 53.28 ? 1 A 1
...
3313 O O . HOH K 5 . ? 75.025 34.314 45.405 1 28.02 ? 454 A 1
3314 O O . HOH K 5 . ? 72.491 43.615 41.507 1 7.99 ? 455 A 1
3315 O O . HOH K 5 . ? 74.729 45.2 40.724 1 6.87 ? 456 A 1
他の書き込み方法や、PDB・mmJSONへの書き込みについてはドキュメントをご確認ください。
SMCRAデータ構造
GEMMIでは、gemmi.read_structure()を使って構造データを Structure-Model-Chain-Residue-Atom (SMCRA) フレームワークとして読み込むことができます。このデータ構造は、Bio.PDBのSMCRAと同様の概念であり、階層的な分子構造の解析を可能にします。
GEMMIを使ったタンパク質構造情報の操作
GEMMIを使ったタンパク質構造情報の操作感は、Bio.PDBのそれとほとんど同じです。
ただ、GEMMIの方がCIFファイルにより適応した使用感になっています。
SMCRAの階層構造
Modelが複数あるシニョリンのNMRエントリーである1uao を使ってみます
import gemmi
st = gemmi.read_structure('1uao.cif.gz')
print(st.name, type(st)) # structure
for model in st:
print('\t', model.num, type(model)) # model
for chain in model:
print('\t\t', chain.name, type(chain)) # chain
for i,residue in enumerate(chain):
if i == 0:
print('\t\t\t', residue.name, type(residue)) # residue
for j,atom in enumerate(residue):
if j == 0:
print('\t\t\t\t', atom.name, type(atom)) # atom
1UAO <class 'gemmi.Structure'>
1 <class 'gemmi.Model'>
A <class 'gemmi.Chain'>
GLY <class 'gemmi.Residue'>
N <class 'gemmi.Atom'>
2 <class 'gemmi.Model'>
A <class 'gemmi.Chain'>
GLY <class 'gemmi.Residue'>
N <class 'gemmi.Atom'>
3 <class 'gemmi.Model'>
A <class 'gemmi.Chain'>
GLY <class 'gemmi.Residue'>
N <class 'gemmi.Atom'>
4 <class 'gemmi.Model'>
A <class 'gemmi.Chain'>
GLY <class 'gemmi.Residue'>
N <class 'gemmi.Atom'>
...
18 <class 'gemmi.Model'>
A <class 'gemmi.Chain'>
GLY <class 'gemmi.Residue'>
N <class 'gemmi.Atom'>
階層構造になっているのがわかりますね。
GEMMIにおけるSMCRA構造の基本操作
SMCRA構造では各階層で共通した操作感で操作することが可能です。これ以降の内容を理解するために、先に共通する基本的な操作を確認しておきましょう。
name プロパティ .name
対象クラス:全て(ただしMoldelは.num)
対象外クラス:なし
structure.nameでそのオブジェクトのnameを確認できます。
ただし、Modelに関しては.numになっています(GEMMI 0.7.0~)。
st = gemmi.read_structure('1alk.cif')
model = st[0]
chain = model[0]
residue = chain[0]
atom = residue[0]
print('Structure', st.name)
print('Model', model.num, type(model.num))
print('Chain', chain.name)
print('Residue', residue.name)
print('Atom', atom.name)
Structure 1ALK
Model 1 <class 'int'>
Chain A
Residue THR
Atom N
ちなみに各name(num)はCIFファイル内の以下の情報から抽出しているようです。
| クラス | メソッド名 | カテゴリ | タグ | 例 |
|---|---|---|---|---|
Structure |
name |
data_XXXX | - | data_1ALK -> '1ALK'
|
Model |
num |
_atom_site | pdbx_PDB_model_num | 1 -> 1 (int型) |
Chain |
name |
_atom_site | auth_asym_id | A -> 'A'
|
Residue |
name |
_atom_site | auth_comp_id | THR -> 'THR'
|
Atom |
name |
_atom_site | auth_atom_id | CA -> 'CA'
|
0-based index __getitem__(index:int) __delitem__(index:int)
対象クラス: Structure Model Chain Residue
対象外クラス:Atom
__getitem__(index:int)はAtomを除いた階層に定義されており、list型のようにstructure[0]で下層のクラスにindex(0-based)を使って下層のオブジェクトを取り出すことができます。もちろん、structure[-1]のように負の値を使って逆順で操作することも可能です。
また同様に__delitem__(index:int)も定義されているため、del structure[0]でindexに対応する下層のオブジェクトを削除できます。
st = gemmi.read_structure('1uao.cif.gz')
# 取り出し
print(st[0]) # 0 始まりであることに注意
print(st[2])
print(st[-1]) # listと同様に負の値も扱える
# 削除
del st[0] # 先頭のModelを削除
print(st[0])
<gemmi.Model 1 with 1 chain(s)>
<gemmi.Model 3 with 1 chain(s)>
<gemmi.Model 18 with 1 chain(s)>
<gemmi.Model 2 with 1 chain(s)>
>>> st = gemmi.read_structure('1alk.cif.gz')
# 先頭のModelの2番目のChainの11番目のResidueの3番目のAtom
>>> st[0][1][10][2]
<gemmi.Atom C at (82.2, 24.3, 23.4)>
あくまで 0-based index であり、ファイル内についている番号とは一致しません。つまり残基番号などに対応しているわけではないので、勘違いしないようにしましょう。
残基番号が1から始まっていない場合や、Chainの途中に欠損がある場合などは要注意です。
ここでいう残基番号とはCIFファイル内の**_site_atom.auth_seq_id**のことを指しています。
>>> st = gemmi.read_structure('1alk.cif.gz')
>>> st[0][0][1] # index=1はchainの二番目の残基
<gemmi.Residue 2(PRO) with 7 atoms>
>>> st = gemmi.read_structure('5za2.cif.gz')
>>> st[0][0][0]
<gemmi.Residue 8(LEU) with 8 atoms> # 残基番号が1から始まるとも限らない
残基番号で残基にアクセスする方法に関しては次のnameを使った操作と本記事の残基番号でresidueへアクセスするを参照ください。
nameを使った操作 __getitem__(name:str)
対象クラス: Model Chain Residue
対象外クラス: Structure Atom
Structure Atom以外では0-based indexの代わりに文字列型のnameで下層にアクセスすることもできます。これによって、ファイル内に書かれているChainIDや残基番号、AtomIDでアクセスすることができます。
しかし、chain['残基番号']では返されるのはResidueではなく、代わりにResidueGroupが返されます。(residue['atom_id']の場合も同様でAtomGroupが返される)。
これらの他、chain['残基番号']とresidue['atom_id']には注意しなければならない点があるので各項の説明(residue / atom)をご覧ください。
st = gemmi.read_structure('1alk.cif.gz')
model = st[0] # Structureは__getitem__(name:str)を持たないので注意
chian_a = model['A']
print(chian_a)
residue_10 = chian_a['10']
print(residue_10) # ResidueではなくResidueGroup
residue_10 = residue_10[0] # Residueに変換(説明省略)
print(residue_10)
atom_ca = residue_10['CA'] # Cα原子
print(atom_ca) # AtomではなくAtomGroup
atom_ca = atom_ca[0] # Atomに変換(説明省略)
print(atom_ca)
<gemmi.Chain A with 456 res>
<gemmi.ResidueGroup [10(ARG)]>
10(ARG)
<gemmi.AtomGroup CA, sites: 1>
<gemmi.Atom CA at (54.4, 41.4, 19.2)>
__delitem__(name:str)はModel以外できないので注意(Modelもやらない方が良さそう)
イテレータ __iter__
対象クラス: Structure Model Chain Residue
対象外クラス:Atom
Atomを除いたすべての階層で__iter__が定義されています。そのため、for文等で下層のクラスを再帰的に呼び出すことができます。前項のSMCRAデータ構造で階層を確認したコードもこの機能を使って書いています。
structure = gemmi.read_structure('file_path')
for model in st:
# Modelに関する処理
for chain in model:
# Chainに関する処理
for residue in chain:
# Residueに関する処理
for atom in residue:
# Atomに関する処理
import gemmi
chainA = gemmi.read_structure('1uao.cif.gz')[0]['A']
residues = []
for residue in chainA: # for文でChainを処理
residues.append(residue.name)
print(residues)
# もちろんリスト内包表記も使える
residues = [residue.name for residue in chainA]
print(residues)
['GLY', 'TYR', 'ASP', 'PRO', 'GLU', 'THR', 'GLY', 'THR', 'TRP', 'GLY']
['GLY', 'TYR', 'ASP', 'PRO', 'GLU', 'THR', 'GLY', 'THR', 'TRP', 'GLY']
またChainクラスのみにおいてsliceを使って操作が可能です。
chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
for residue in chain[:10]: # 10残基まで
print(residue, end=' ')
1(THR) 2(PRO) 3(GLU) 4(MET) 5(PRO) 6(VAL) 7(LEU) 8(GLU) 9(ASN) 10(ARG)
length __len__
対象クラス: Structure Model Chain Residue
対象外クラス:Atom
Atomを除いたSMCRのクラスには__len__が定義されているため、len(structure)などで下層のオブジェクトの数を確認できます。
st = gemmi.read_structure('1uao.cif.gz')
model = st[0]
chain = model[0]
residue = chain[0]
print('Model length:', len(model))
print('Chain length:', len(chain))
print('Residue length:', len(residue))
Model length: 1
Chain length: 10
Residue length: 9
altlocなどの関係から必ずしも残基数や原子数に対応しているわけではないことに注意!!
下層オブジェクトの追加 add_***
対象クラス: Structure Model Chain Residue
対象外クラス:Atom
例えば、**「Chainオブジェクトに新しいResidueオブジェクトを追加する」**などといった既存のオブジェクトに下層のオブジェクトを追加する際は、add_***()(***はmodelなど)メソッドを使用します。
| メソッド | 追加対象 | 追加オブジェクト | オプション | 説明 |
|---|---|---|---|---|
structure.add_model(model) |
Structure |
Model |
pos: int = -1 |
posで挿入位置を指定(デフォルトは末尾-1)以下のクラスも同様。 |
model.add_chain(chain) |
Model |
Chain |
pos: int = -1,unique_name: bool = False
|
unique_nameで名前の重複回避を指定可能(デフォルトはFalse)。 |
chain.add_residue(residue) |
Chain |
Residue |
pos: int = -1 |
|
residue.add_atom(atom) |
Residue |
Atom |
pos: int = -1 |
# StructureにModelを追加
st = gemmi.read_structure('1uao.cif.gz')
print(st[1], end=' -> ')
st.add_model(gemmi.Model(777), pos=1)
print(st[1])
# ModelにChainを追加
model = gemmi.read_structure('1uao.cif.gz')[0]
print(model[0], end=' -> ')
model.add_chain(gemmi.Chain('NEW'), pos=0)
print(model[0])
del model[0] # 一旦削除
# unique_name=True
print(list(model), end=' -> ')
model.add_chain(gemmi.Chain('A'), unique_name=True) # unique_name=Trueで同名のChainを追加
print(list(model))
# ChainにResidueを追加
chain = gemmi.read_structure('1uao.cif.gz')[0][0]
print(chain[-2], end=' -> ')
chain.add_residue(gemmi.Residue(), pos=-2)
print(chain[-2])
# ResidueにAtomを追加
residue = gemmi.read_structure('1uao.cif.gz')[0][0][0]
print(residue[1], end=' -> ')
residue.add_atom(gemmi.Atom(), pos=1)
print(residue[1])
<gemmi.Model 2 with 1 chain(s)> -> <gemmi.Model 777 with 0 chain(s)>
<gemmi.Chain A with 10 res> -> <gemmi.Chain NEW with 0 res>
[<gemmi.Chain A with 10 res>] -> [<gemmi.Chain A with 10 res>, <gemmi.Chain B with 0 res>]
9(TRP) -> 10(GLY)
<gemmi.Atom CA at (-6.9, -0.7, 2.9)> -> <gemmi.Atom at (0.0, 0.0, 0.0)>
モデルを追加・削除すると参照が無効になる場合があります
下層のオブジェクトを追加・削除すると、同じオブジェクトに含まれる他のモデルを指す「参照」や「ポインタ」が使えなくなることがあります。これは、内部実装にC++の std::vector を用いており、要素(モデルなど)の追加・削除時にメモリが再配置(re-allocation)される可能性があるためです。
-
st.add_model(...)は、すべてのモデルへの参照を無効化する恐れがあります -
del st[i](__delitem__) は、削除した位置より後にあるモデルへの参照を無効化します
したがって、次のようなコードを書くときは注意が必要です:
model_reference = st[0]
st.add_model(...) # ここで model_reference が無効になる可能性
model_reference = st[0] # 改めて有効な参照を取り直す
上記のように、追加・削除の前後で参照を取り直すことが必要です。
同じことは削除(__delitem__)を行った場合にも当てはまる点にご注意ください。
オブジェクトをコピーする clone()
対象クラス:全て
対象外クラス:なし
SMCRAオブジェクトをコピーしたい場合はclone()を使用します。structure_1 = structure_2のようにただ単に別のオブジェクトに代入するだけではコピーにならないので注意してください。
# Structure
st = gemmi.read_structure('1uao.cif.gz')
st_1 = st # 代入
st_c = st.clone() # clone()
# Model
model = st[0]
model_1 = model
model_c = model.clone()
# Chain
chain = model[0]
chain_1 = chain
chain_c = chain.clone()
# Residue
residue = chain[0]
residue_1 = residue
residue_c = residue.clone()
# Atom
atom = residue[0]
atom_1 = atom
atom_c = atom.clone()
# 各オブジェクトのIDを表示
print('Structure:', id(st), id(st_1), id(st_c))
print('Model:', id(model), id(model_1), id(model_c))
print('Chain:', id(chain), id(chain_1), id(chain_c))
print('Residue:', id(residue), id(residue_1), id(residue_c))
print('Atom:', id(atom), id(atom_1), id(atom_c))
Structure: 5095774704 5095774704 5095768528
Model: 5095771472 5095771472 5095773360
Chain: 5095769072 5095769072 5095771248
Residue: 5095770384 5095770384 5095774192
Atom: 5094541936 5094541936 5094552304
編集前のオブジェクトを残しておきたいときなどにclone()を活用しましょう
st = gemmi.read_structure('1alk.cif.gz')
st_c = st.clone()
st.add_model(gemmi.Model(777))
print('st:', st, 'length:', len(st))
print('st_c:', st_c, 'length:', len(st_c))
st: <gemmi.Structure 1ALK with 2 model(s)> length: 2
st_c: <gemmi.Structure 1ALK with 1 model(s)> length: 1
Structure
ここからは各SMCRAのクラスについて詳細にみていきます。
まず、SMCRAの最上位クラス、Structureクラスについて見てみましょう。Structureのオブジェクトには、1つ以上のModelが格納されています。
主にNMRで撮られたエントリーなどの場合にModelが複数含まれます、PDBから取得した構造の場合、NMR以外は基本的に一つのModelしか含まれていないことが多いです。
>>> st = gemmi.read_structure('1alk.cif.gz')
>>> st
<gemmi.Structure 1ALK with 1 model(s)>
>>> st.info['_exptl.method'] # 構造決定手法の確認
'X-RAY DIFFRACTION'
>>> len(st)
1
# NMRの構造の場合
>>> st = gemmi.read_structure('1uao.cif.gz')
>>> st # NMRなどの場合複数モデルが含まれる
<gemmi.Structure 1UAO with 18 model(s)>
>>> st.info['_exptl.method']
'SOLUTION NMR'
>>> len(st)
18
Structure から Model へのアクセス
StrucutureオブジェクトからModelを取り出す方法は0-based indexとイテレータの二種類があります。
以下のコードは基本操作で扱ったものと同様なのでそちらを読んだ方は読み飛ばしてもOKです。
>>> st = gemmi.read_structure('1uao.cif.gz')
>>> st[0] # 先頭のモデル
<gemmi.Model 1 with 1 chain(s)>
>>> st[1] # 二番目のモデル
<gemmi.Model 2 with 1 chain(s)>
>>> st[-1] # 最後尾のモデル
<gemmi.Model 18 with 1 chain(s)>
>>> st = gemmi.read_structure('1uao.cif.gz')
>>> len(st)
18
>>> for model in st: # __iter__を使って再帰的にmodelを取得
print(model)
<gemmi.Model 1 with 1 chain(s)>
<gemmi.Model 2 with 1 chain(s)>
<gemmi.Model 3 with 1 chain(s)>
<gemmi.Model 4 with 1 chain(s)>
...
<gemmi.Model 18 with 1 chain(s)>
# モデル番号とindexはからなずしも一致しない
>>> st = gemmi.read_structure('1uao.cif.gz')
>>> print(0, st[0].num) # idexとモデル番号
0 1
version 0.6.7までは__get_item__(name:str)が定義されており、モデル番号を使ってアクセスできましたが、0.7.0以降は削除されたようです(Chain Residueは可能)。
>>> import gemmi
>>> gemmi.__version__
0.6.7
>>> print(0, st[0].name) # 0.6.7まではnameだった
0, 1
>>> st['1'] # __get_item__(name:str)
<gemmi.Model 1 with 1 chain(s)>
>>> import gemmi
>>> gemmi.__version__
>>> st = gemmi.read_structure('1uao.cif.gz')
>>> st['1'] # __get_item__(name:str)は定義されていないが使用はできる
<gemmi.Model 2 with 1 chain(s)> # モデル番号1ではなく2が取り出される(indexの時と同じ)
Structure に Model を 追加/削除 する
StrucutureオブジェクトからModelを削除するには0-based indexを使って行います。
基本操作で説明した以外のものとしてsliceを使った削除(__delitem__(arg:slice))があります。
>>> st = gemmi.read_structure('1uao.cif.gz')
>>> len(st) # Modelの数を確認
18
>>> del st[1:] # 先頭以降のモデルを全部削除(__delitem__(arg:slice))
>>> len(st)
1
また、structure.renumber_models()メソッドを呼び出すことで、複数モデルをまとめた後、1から順番にモデル番号(Model.num)を自動的に振り直してくれます。
st = gemmi.read_structure('1uao.cif.gz')
st.add_model(gemmi.Model(777), pos=5) # 5番目にModel(777)を追加
print('bfore:', end=' ')
for model in st:
print(model.num, end=' ')
print()
print('after:', end=' ')
st.renumber_models() # Modelの番号を振り直す
for model in st:
print(model.num, end=' ')
bfore: 1 2 3 4 5 777 6 7 8 9 10 11 12 13 14 15 16 17 18
after: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Properties
StructureにはModelを格納しているだけでなく、いくつかのプロパティも所持しており、resolutionやヘッダーに書かれた基本情報にアクセスすることができます。
| Property | データ型 | 説明 |
|---|---|---|
name |
str |
通常はファイルのベース名またはPDBコード |
cell |
UnitCell |
ユニットセル |
spacegroup_hm |
str |
ヘルマン・モーガン記法による完全な空間群名(通常は座標ファイルから取得) |
ncs |
NcsOpList |
NCS操作のリスト。通常はMTRIX記録またはstruct_ncs_operカテゴリから取得 |
resolution |
float |
REMARK 2または3からの分解能値 |
entities |
EntityList |
subchainに関する追加情報。エンティティタイプやポリマーの配列など |
connections |
ConnectionList |
mmCIFのstruct_connカテゴリ、またはPDBのLINKおよびSSBOND記録に対応する接続のリスト |
assemblies |
AssemblyList |
PDBのREMARK 350または対応するmmCIFカテゴリで定義された生物学的アセンブリのリスト |
input_format |
CoorFormat |
読み込まれたファイル形式 |
has_d_fraction |
bool |
重水素の表現方法 |
origx |
Transform |
PDBのORIGX記録またはmmCIFのdatabase_pdb_matrix.origxからの行列 |
info |
InfoMap |
キーがmmCIFタグである最小限のメタデータ |
meta |
Metadata |
構造化されたメタデータ。異なるmmCIFタグに対応するほぼ100のプロパティ |
raw_remarks |
RawRemarksList |
PDBファイルからのREMARK記録。入力ファイルが異なる形式の場合は空 |
cispeps |
CisPepList |
PDBのCISPEP記録またはmmCIFのstruct_mon_prot_cisカテゴリに対応 |
mod_residues |
ModResList |
修飾された残基のリスト |
helices |
HelixList |
PDBのHELIX記録またはmmCIFのstruct_confカテゴリに対応 |
sheets |
SheetList |
PDBのSHEET記録またはmmCIFのstruct_sheet_rangeカテゴリに対応 |
shortened_ccd_codes |
ShortenedCcdCodeList |
短縮されたCCDコードと元のコードの対応リスト |
流石にこれら全部を扱うのは難しいので以下に一部だけ紹介します。
import gemmi
>>> st = gemmi.read_structure('1alk.cif.gz')
# name/filename
>>> st.name
'1ALK'
# resolution
>>> st.resolution #float
2.0
# 基本情報 info
>>> info = st.info
>>> print('type:', type(info))
>>> for key, value in info.items():
print(key, value)
type: <class 'gemmi.InfoMap'>
_cell.Z_PDB 16
_entry.id 1ALK
_exptl.method X-RAY DIFFRACTION
_pdbx_database_status.recvd_initial_deposition_date 1993-03-03
_struct.title REACTION MECHANISM OF ALKALINE PHOSPHATASE BASED ON CRYSTAL STRUCTURES. TWO METAL ION CATALYSIS
_struct_keywords.pdbx_keywords ALKALINE PHOSPHATASE
_struct_keywords.text ALKALINE PHOSPHATASE
# メタデータ meta
>>> meta = st.meta
>>> type(meta)
gemmi.Metadata
>>> meta.authors
['Kim, E.E.', 'Wyckoff, W.']
>>> meta.refinement[0].resolution_high
2.0
>>> meta.software[0].name
X-PLOR
そのほかのpropertiesを確認したい方はドキュメントをご確認ください。
Methods
Structureクラスには多くの便利なメソッドが定義されています。
| メソッド名 | 引数 | 返り値の型 | 説明 |
|---|---|---|---|
clone |
- | Structure |
Structureオブジェクトをコピーします。 |
remove_alternative_conformations |
- | None |
構造から代替コンフォメーションを取り除きます。(破壊的メソッド) |
remove_empty_chains |
- | None |
構造から空のチェーンを取り除きます。(破壊的メソッド) |
remove_hydrogens |
- | None |
構造から水素原子を取り除きます。(破壊的メソッド) |
remove_ligands_and_waters |
- | None |
構造からリガンドおよび水分子を取り除きます。(破壊的メソッド) |
remove_waters |
- | None |
構造から水分子を取り除きます。(破壊的メソッド) |
make_mmcif_document |
groups: MmcifOutputGroups = MmcifOutputGroups(True) |
gemmi.cif.Document |
現在のStructureからgemmi.cif.Documentを作成します。 |
make_mmcif_block |
groups: MmcifOutputGroups = MmcifOutputGroups(True) |
gemmi.cif.Block |
現在のStructureからgemmi.cif.Blockを作成します。 |
add_model |
model: Model, pos: int = -1 |
Model |
現在のStructureに任意のモデルオブジェクトを追加します。 |
renumber_models |
- | None |
モデル番号(Model.num)を自動的に振り直します。 |
add_entity_types |
overwrite: bool = False |
None |
エンティティタイプの情報を追加します。 |
add_entity_ids |
overwrite: bool = False |
None |
エンティティIDの情報を追加します。 |
add_conect |
serial1: int, serial2: int, order: int |
None |
CONECT記録を追加します。 |
clear_conect |
- | None |
CONECT記録をクリアします。 |
add_connection |
connection: Connection |
None |
構造に接続情報を追加します。 |
assign_subchains |
force: bool = False, fail_if_unknown: bool = True |
None |
サブチェーンを割り当てます。 |
ensure_entities |
- | None |
必要なエンティティを追加します。 |
deduplicate_entities |
- | None |
重複するエンティティを削除します。 |
setup_entities |
- | None |
エンティティ情報を設定します。 |
write_pdb |
path: str, options: PdbWriteOptions = PdbWriteOptions() |
None |
現在の構造をPDB形式で書き込みます。 |
shorten_ccd_codes |
- | None |
5文字のCCDコードを短縮します。 |
shorten_chain_names |
- | None |
長いChainIDを短縮します。 |
transform_to_assembly |
`assembly_name: str, how: HowToNameCopiedChain, logging: object | None = None, keep_spacegroup: bool = False, merge_dist: float = 0.2` | None |
多いので詳細などは省略しますが、remove_***は前処理等でよく行う、水分子の削除等が簡単に行えるので便利です。また、make_mmcif_document/blockは構造ベースから構文ベースの編集に移行する時に使用します。CIFファイルへの書き込みはこのメソッドを使って、一度Document/Blockにする必要があります (構造の書き込み)。
Model
Structureの次の階層であるModelは Chain オブジェクトを保持しています。
>>> st = gemmi.read_structure('1alk.cif.gz')
>>> model = st[0] # 先頭のModelを取得
>>> model
<gemmi.Model 1 with 2 chain(s)>
>>> len(model) # len(Model)で保持されているChainの数を確認できる
2
Model から Chain へのアクセス
以下の内容は基本操作で扱った内容と同様です。
1alkだとChainが2本しかないのでChainが6本の5fqdを使ってみましょう。
>>> model = gemmi.read_structure('5fqd.cif.gz')[0]
>>> for chain in model: # __iter__
print(chain)
<gemmi.Chain A with 843 res>
<gemmi.Chain B with 435 res>
<gemmi.Chain C with 342 res>
<gemmi.Chain D with 883 res>
<gemmi.Chain E with 379 res>
<gemmi.Chain F with 342 res>
>>> model[0] # 0-based index __get_item__(index:int)
<gemmi.Chain A with 843 res>
>>>> model['A'] # ChainID(名)でもアクセス可能 __get_item__(name:str)
<gemmi.Chain A with 843 res>
Structureと異なるのは__get_item__(name:str)が定義されているためChainID(名)でもアクセス可能な点です。
Model に Chain を 追加/削除 する
以下の内容は基本操作で扱った内容と同様です。
他のクラスのadd_chainと異なる点は、引数unique_name=Trueを指定すると、同名のチェーンが既にあったときに自動でチェーン名を変えて衝突を回避してくれます。
>>> model = gemmi.read_structure('1alk.cif.gz')[0]
>>> model.add_chain(gemmi.Chain('C'))
>>> list(model)
[<gemmi.Chain A with 456 res>,
<gemmi.Chain B with 456 res>,
<gemmi.Chain C with 0 res>]
# unique_name=Trueの場合
>>> model = gemmi.read_structure('1alk.cif.gz')[0]
>>> model.add_chain(gemmi.Chain('A'), unique_name=True)
>>> list(model)
[<gemmi.Chain A with 456 res>,
<gemmi.Chain B with 456 res>,
<gemmi.Chain C with 0 res>] # 勝手にCにしてくれている
削除はidenxの他、del model[name:str]やremove_chain(name:str)でも可能です。
Properties
Structureと異なりModelにはnum:int(モデル番号)しかプロパティがありません。
version 0.6.7 までは num:intではなくname:strとして定義されていたので注意
Methods
Structure同様にModelにも多数のメソッドが備わっています。
| メソッド名 | 引数 | 返り値の型 | 説明 |
|---|---|---|---|
all |
- | CraGenerator |
モデル内のすべてのCRAを生成します。 |
get_subchain |
name: str |
ResidueSpan |
指定した名前のサブチェーンを取得します。 |
subchains |
- | list[ResidueSpan] |
モデル内のすべてのサブチェーンを返します。 |
find_residue_group |
chain: str, seqid: SeqId |
ResidueGroup |
指定したチェーンとシーケンスIDに対応するResidueGroupを取得します。 |
sole_residue |
chain: str, seqid: SeqId |
Residue |
指定したチェーンとシーケンスIDに対応する単一のResidueを取得します。 |
get_all_residue_names |
- | list[str] |
モデル内のすべてのユニークな残基名を返します。 |
find_cra |
arg0: AtomAddress, ignore_segment: bool = False |
CRA |
指定されたAtomAddressに対応するCRAを検索します。 |
find_chain |
name: str |
Chain |
指定された名前の最初のチェーンを返します。 |
find_last_chain |
name: str |
Chain |
指定された名前の最後のチェーンを返します。 |
add_chain |
chain: Chain, pos: int = -1, unique_name: bool = False または name: str
|
Chain |
チェーンを追加します。 |
remove_chain |
name: str |
None |
指定された名前のチェーンを削除します。 |
remove_alternative_conformations |
- | None |
代替コンフォメーションを削除します。 |
remove_hydrogens |
- | None |
水素原子を削除します。 |
remove_waters |
- | None |
水分子を削除します。 |
remove_ligands_and_waters |
- | None |
リガンドおよび水分子を削除します。 |
has_hydrogen |
- | bool |
モデル内に水素が含まれているかを判定します。 |
count_atom_sites |
`sel: Selection | None = None` | int |
count_occupancies |
`sel: Selection | None = None` | float |
calculate_mass |
- | float |
モデルの質量を計算します。 |
calculate_center_of_mass |
- | Position |
モデルの質量中心を計算します。 |
calculate_b_iso_range |
- | tuple[float, float] |
B因子の等方性範囲を計算します。 |
calculate_b_aniso_range |
- | tuple[float, float] |
B因子の異方性範囲を計算します。 |
transform_pos_and_adp |
tr: Transform |
None |
モデル内の座標とADPを変換します。 |
split_chains_by_segments |
arg: HowToNameCopiedChain |
None |
チェーンをセグメントごとに分割します。 |
clone |
- | Model |
モデルをコピーします。 |
いくつか代表的なものについて簡単に紹介します。
all
Model内の全Atomを一度に処理したい場合、model.all() というイテレータを使ってアクセスできます。
for cra in model.all():
# cra.chain, cra.residue, cra.atom にアクセス可能
if cra.residue.name == 'MET':
...
ここで CRA(Chain-Residue-Atom)クラスを得られ、対応するChain・Residue・Atom をまとめて取り出せます。
計算系メソッド
>>> model = gemmi.read_structure('1alk.cif.gz')[0]
>>> model.count_atom_sites()
6630
>>> model.count_occupancies()
6630.0
>>> model.calculate_mass()
88096.32512200014
>>> model.calculate_center_of_mass()
<gemmi.Position(64.6643, 30.6177, 28.7573)>
>>> model.calculate_b_iso_range()
(0.0, 63.06999969482422)
>>> model.count_atom_sites(gemmi.Selection('[S]'))
24
Selectionについては今回は割愛します(こちらを参照してください)
Subchain
通常ChianIDといった場合、cifファイルのauth_asym_idを指していることが多く、実際にGEMMIの場合でもChainクラスはauth_asym_idのまとまりを取り扱います。しかし、mmCIFにはlabel_asym_idというもう一つの区分が存在します。GEMMIではこのlabel_asym_idのまとまりをsubchainと呼んでおり、subchainsやget_subchainメソッド等で取得することができます。
auth_asym_idとlabel_asym_idの違いなどについては以下等を参照してください
model = gemmi.read_structure('1alk.cif.gz')[0]
print('Chain (auth_asym_id):', end=' ')
for chain in model:
print(chain.name, end=' ')
print()
print('Subchain (label_asym_id):', end=' ')
for subchain in model.subchains():
print(subchain.subchain_id(), end=' ')
Chain (auth_asym_id): A B
Subchain (label_asym_id): A C D E F K B G H I J L
実はこのsubchainはそれぞれのenityに対応するインスタンスに割り当てられているため、機械的な操作においてはとても有用な場合があります。
Chain
Modelの次の階層であるChainは Residue クラスを保持しています。このChainはmmCIFフォーマットの_atom_site.auth_asym_idに該当します。
>>> st = gemmi.read_structure('1alk.cif.gz')
>>> chain = st[0]['A'] # model 0 の chain A を取得
>>> chain
<gemmi.Chain A with 456 res>
>>> chain.name # nameプロパティでchain名にアクセス
'A'
>>> len(chain) # len(Model)で保持されているResidueの数を確認できる
456
Chain から Residue へのアクセス
基本的な操作はSMCRA構造の基本操作をご確認ください。
Slice
Chainの場合はsliceを使うことができます(__getitem__(arg: slice))。返ってくるのはList[Residue]になります。
>>> chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
>>> chain[5:10] # Chain A のindex 5~10までのResidueを取得
[<gemmi.Residue 6(VAL) with 7 atoms>,
<gemmi.Residue 7(LEU) with 8 atoms>,
<gemmi.Residue 8(GLU) with 9 atoms>,
<gemmi.Residue 9(ASN) with 8 atoms>,
<gemmi.Residue 10(ARG) with 11 atoms>]
>>> type(chain[5:10]) # 型はlist
list
>>> chain[:3] # 先頭〜index 3まで
[<gemmi.Residue 1(THR) with 7 atoms>,
<gemmi.Residue 2(PRO) with 7 atoms>,
<gemmi.Residue 3(GLU) with 9 atoms>]
>>> chain[-5:] # 末尾5~
[<gemmi.Residue 452(MG) with 1 atoms>,
<gemmi.Residue 453(PO4) with 5 atoms>,
<gemmi.Residue 454(HOH) with 1 atoms>,
<gemmi.Residue 455(HOH) with 1 atoms>,
<gemmi.Residue 456(HOH) with 1 atoms>]
残基番号でResidueへアクセスする (ResidueGroup)
残基を参照したい時は0-based indexではなく、論文などに書かれている残基番号でアクセスしたいことが多いと思います。GEMMIでも同様にChainから任意の残基番号にアクセスすることができます。
残基番号でアクセスするときはchain[pdb_seqid:str](__getitem__(pdb_seqid: str))でアクセスしますが、返ってくるのはResidueではなくResidueGroupになります。
>>> chain = gemmi.read_structure('1a00.cif.gz')[0]['A']
>>> chain['1'] # 文字列の残基番号 intだとindexになるので注意
<gemmi.ResidueGroup [1(THR)]>
ResidueGroupから該当残基を出すにはgroup['残基名']で取り出すことができます。
>>> group = chain['1']
>>> group
<gemmi.ResidueGroup [1(THR)]>
>>> group['THR'] # 残基名='THR'
<gemmi.Residue 1(THR) with 7 atoms>
>>> group[0] # 0-based indexでも取得できる
<gemmi.Residue 1(THR) with 7 atoms>
なぜ、直接ResidueではなくResidueGroupなのかというと、同じ残基番号の残基が複数ある場合(microheterogeneitiesなど)があり、それらに対処するためです。
同じ残基番号の残基が複数ある場合
microheterogeneitiesに関しては
のHow is microheterogeneity/polymorphism handledをご覧ください
では実際に、同じ残基番号の残基が複数ある場合もみてみましょう。
microheterogeneityがある場合、_entity_poly_seq.heteroもしくは_pdbx_poly_seq_scheme.heteroに'y'が列挙されます。
7a0lはmicroheterogeneityを持つエントリーになります。確認してみましょう。
import polars as pl
import gemmi
block = gemmi.cif.read('7a0l.cif.gz')[0]
df = pl.DataFrame(block.get_mmcif_category('_pdbx_poly_seq_scheme'),strict=False)
print(
df.filter(pl.col('hetero').eq('y'))
.select([
'pdb_strand_id','seq_id','auth_seq_num', 'mon_id', 'hetero'
])
)
shape: (2, 5)
┌───────────────┬────────┬──────────────┬────────┬────────┐
│ pdb_strand_id ┆ seq_id ┆ auth_seq_num ┆ mon_id ┆ hetero │
│ --- ┆ --- ┆ --- ┆ --- ┆ --- │
│ str ┆ str ┆ str ┆ str ┆ str │
╞═══════════════╪════════╪══════════════╪════════╪════════╡
│ A ┆ 36 ┆ 35 ┆ CYS ┆ y │
│ A ┆ 36 ┆ 35 ┆ CSO ┆ y │
└───────────────┴────────┴──────────────┴────────┴────────┘
(上記コードに関しては こちらの記事 を参照してください)
ご覧の通りChain Aの35番目(auth_seq_id)残基がCYSとCSOの二つ存在しているようです。read_structureで構造を読み込んで、'chain['35']'をみてみましょう。
>>> chain = gemmi.read_structure('7a0l.cif.gz')[0]['A']
>>> chain['35']
<gemmi.ResidueGroup [35(CYS) 35(CSO)]>
>>> print(chain['35'][0], chain['35'][1])
35(CYS) 35(CSO)
>>> print(chain['35']['CYS'], chain['35']['CSO'])
35(CYS) 35(CSO)
読み込んだ構造でも二つの残基(CYS/CSO)になって、ResidueGroupに入っていますね。PDBにはこういった構造がいくつか存在します。プログラムを作る際は事前にこれらをどう扱うかを決めておきましょう。
例: 最初に出てくる残基だけをあつかう chain.first_conformer()
chain = gemmi.read_structure('7a0l.cif.gz')[0]['A']
for res in chain.first_conformer():
if res.seqid.num in [34, 35, 36]:
print(res, end=' ')
34(VAL) 35(CYS) 36(VAL)
first_conformer()で取得されるFirstConformerResは0-based indexでアクセスできません。純粋なIterator[Residue]として機能します。
Insertion code (icode)
実は残基番号での残基へのアクセスの仕方は正確にはchain[残基番号:str]ではありません。
正確にはchain[残基番号+icode(insertion code)]でアクセスする必要があります。
例えば 1igy のChainBの 残基番号82は四つの残基が含まれています
import polras
from gemmi import cif
block = cif.read('1igy.cif').sole_block()
df = pl.DataFrame(
block.get_mmcif_category('_atom_site'),
strict=False
)
print(df.unique(['label_seq_id', 'auth_asym_id']
).filter(
pl.col('auth_seq_id') == '82',
pl.col('auth_asym_id') == 'B'
).select([
'label_seq_id',
'auth_asym_id',
'auth_seq_id',
'pdbx_PDB_ins_code',
'auth_comp_id'
]).sort('label_seq_id'))
shape: (4, 5)
┌──────────────┬──────────────┬─────────────┬───────────────────┬──────────────┐
│ label_seq_id ┆ auth_asym_id ┆ auth_seq_id ┆ pdbx_PDB_ins_code ┆ auth_comp_id │
│ --- ┆ --- ┆ --- ┆ --- ┆ --- │
│ str ┆ str ┆ str ┆ str ┆ str │
╞══════════════╪══════════════╪═════════════╪═══════════════════╪══════════════╡
│ 84 ┆ B ┆ 82 ┆ B ┆ SER │
│ 85 ┆ B ┆ 82 ┆ C ┆ LEU │
│ 83 ┆ B ┆ 82 ┆ A ┆ SER │
│ 82 ┆ B ┆ 82 ┆ null ┆ LEU │
└──────────────┴──────────────┴─────────────┴───────────────────┴──────────────┘
(上記コードに関しては こちらの記事 を参照してください)
実際の構造

これは参照配列(例えばUniProtや論文中の配列番号)に対して実際の蛋白質配列に挿入がある場合の対処として導入されています。これらはCDRなどで頻繁にみられます。またその他にも糖鎖などの修飾残基や溶媒などにおいてもしばしばみられます。
詳しいことは以下を参照してください。
糖鎖の場合は別のChainIDが割り当てられていることも多いです。
また、AlphaFoldの予測構造の場合、microheterogeneityやicodeはないので気にせず処理してもらって問題ありません。
これらを見分けるためにGEMMIではseqid+icodeが必要になります。
>>> st = gemmi.read_structure('1igy.cif')
>>> st[0]['B']['82'] # 82の場合
<gemmi.ResidueGroup [82(LEU)]>
>>> st[0]['B']['82B'] # 82Bの場合
<gemmi.ResidueGroup [82B(SER)]>
>>> st[0]['B']['82 '] # icodeの分を半角スペース一個分空けて指定することで icodeがないことをわかりやすくすることもできる
<gemmi.ResidueGroup [82(LEU)]> # '82'で指定した場合と同じ
プログラム書く際はicodeの有無に注意してコーディングしましょう。
Chain に Residue を 追加/削除 する
基本的な操作はSMCRA構造の基本操作をご確認ください。
Slice
__delitem__(arg:slice)が利用可能です。一方で、chain['残基番号']のようにdel chain['残基番号']は利用できません。
>>> chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
>>> len(chain)
456
>>> del chain[-5:]
>>> len(chain)
451
# __delitem__(pdb_seqid:str)はできない
>>> chain = gemmi.read_structure('1igy.cif')[0]['B']
>>> print(chain['82B'][0], chain[83])
>>> del chain['82B'] # TypeError
Methods
| メソッド名 | 引数 | 返り値の型 | 説明 |
|---|---|---|---|
add_residue |
residue: Residue, pos: int = -1 |
Residue |
残基を追加します。 |
subchains |
- | list[ResidueSpan] |
SubchainをResidueSpanのリストとして返します。 |
whole |
- | ResidueSpan |
Chain全体を表すResidueSpanを返します。 |
get_polymer |
- | ResidueSpan |
polymerを表すResidueSpanを返します。 |
get_ligands |
- | ResidueSpan |
リガンドを表すResidueSpanを返します。 |
get_waters |
- | ResidueSpan |
水分子を表すResidueSpanを返します。 |
get_subchain |
arg: str |
ResidueSpan |
指定したSubchainを表すResidueSpanを返します。 |
previous_residue |
arg: Residue |
Residue |
指定した残基の前の残基を返します。 |
next_residue |
arg: Residue |
Residue |
指定した残基の次の残基を返します。 |
append_residues |
new_residues: Sequence[Residue], min_sep: int = 0 |
None |
残基をまとめてChainに追加します。 |
count_atom_sites |
sel: Selection | None = None |
int |
選択された原子サイトの数をカウントします。 |
count_occupancies |
sel: Selection | None = None |
float |
選択された占有率の合計を計算します。 |
calculate_mass |
- | float |
Chain全体の質量を計算します。 |
calculate_center_of_mass |
- | Position |
Chainの質量中心を計算します。 |
trim_to_alanine |
- | None |
ポリペプチド鎖をポリアラニンに変化させる。 |
first_conformer |
- | FirstConformerRes |
最初のコンフォーマを含む新しいオブジェクトを返します(代替構造を無視します)。 |
clone |
- | Chain |
チェーンをコピーします。 |
chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
print(chain.count_atom_sites())
print(chain.count_occupancies())
print(chain.calculate_mass())
print(chain.calculate_center_of_mass())
3315
3315.0
44048.16256100007
<gemmi.Position(72.3348, 45.2708, 34.6821)>
get_polymer()やwhole()などは次のResidueSpanで説明します。
trim_to_alanine()はなぜかうまく機能しません (ver. 0.7.0)
ResidueSpan
Chainのメソッドの説明に移る前に、Chainに似たオブジェクトであるResidueSpanを紹介します。
ResidueSpanはchain内の連続した複数の残基の領域を“そのまま”取り出せる軽量オブジェクトであり、実際の残基データをコピーせずに扱うことができます。
通常、Chain内にはポリペプチド鎖などのポリマー分子だけでなく、リガンドや水分子なども含まれています。
例えば、あるエントリーのChain Aにタンパク質のペプチド鎖、リガンド、水分子が混在していたとします。タンパク質単体の解析をしたい場合、リガンドや水分子は解析対象外となるため、それらを除外する必要があります。
このようなケースでは、Chainのメソッドであるget_polymer()やget_subchain()を使用することで、扱いたい分子鎖を簡単に切り出して操作することが可能です。
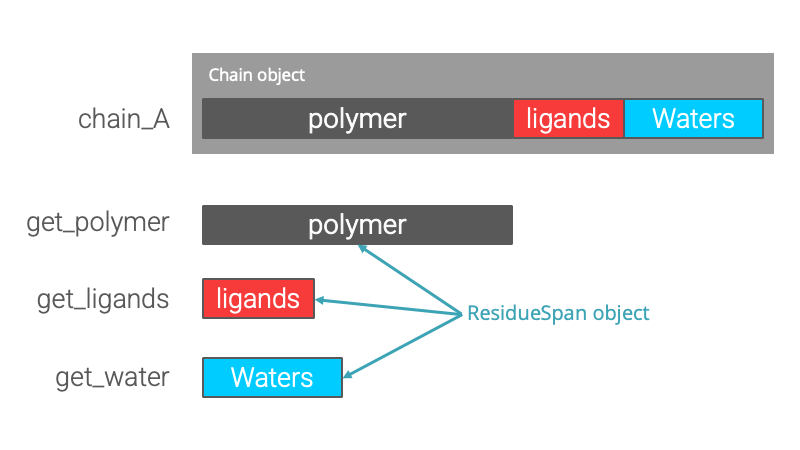
このように、ResidueSpanを活用することで、必要なデータのみを効率的に扱えるようになります。
ResidueSpanの基本操作
また、ResidueSpanからResidueを取り出す基本的な操作はChainとほとんど変わりません。(ただし、sliceは使用できません。)
>>> chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
>>> polymer = chain.get_polymer() # get_polymerについては後述
>>> polymer
<gemmi.ResidueSpan of 449: A [1(THR) 2(PRO) 3(GLU) ... 449(LYS)]>
# 残基にアクセス
>>> polymer[0] # 0-based index
<gemmi.Residue 1(THR) with 7 atoms>
>>> polymer['1'] # pdb_seqid: str
<gemmi.ResidueGroup [1(THR)]>
# sliceは使用できない
# >>> for res in polymer[:5]: # TypeError
# print(res)
# __iter__
>>> len(polymer)
449
>>> for res in polymer:
print(res)
1(THR)
2(PRO)
3(GLU)
...
449(LYS)
残基の追加・削除
Chain 同様にResidueSpan.add_residue(...), del ResidueSpan[i] などで操作可能です
ResidueSpanの__len__()とlength()の違い
ResidueSpanには長さを取得する二つの方法があります:
-
len(ResidueSpan)(__len__)-
意味:
ResidueSpan内の全ての残基数を返します。
-
意味:
-
ResidueSpan.length()- 意味: microheterogeneityを除いたポリマーの実質的な長さを返します。
これらの違いをchainAにmicroheterogeneityを持つエントリー(7A0l)で確認してみましょう。
>>> chain = gemmi.read_structure('7a0l.cif.gz')[0]['A']
>>> polymer = chain.get_polymer()
>>> print(len(polymer), polymer.length())
296 295
- この例では、
len(polymer)が296と出力され、これはすべての残基を含む長さを示しています。 - 一方、
polymer.length()が295で、これはmicroheterogeneityを除外した実質的なポリマーの長さを表しています。
これにより、解析の目的に応じて適切な方法を選択することができます。例えば、ポリマーの正確なサイズを測定する場合にはlength()が便利です。
ChainからResidueSpanを取得するメソッド
get_polymer(), get_ligands(), get_waters()
通常、ポリマー(主鎖)→リガンド→水 の順番で並んでいるという慣習を利用し、get_polymer get_ligands get_waters の3つのメソッドで連続した領域(ResidueSpan)を取得できます。
>>> chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
>>> print(chain.get_polymer())
<gemmi.ResidueSpan of 449: A [1(THR) 2(PRO) 3(GLU) ... 449(LYS)]>
>>> print(chain.get_ligands())
<gemmi.ResidueSpan of 4: C - F [450(ZN) 451(ZN) 452(MG) 453(PO4)]>
>>> print(chain.get_waters())
<gemmi.ResidueSpan of 3: K [454(HOH) 455(HOH) 456(HOH)]>
whole()
whole() を呼ぶと、チェーン内の全残基を1つの ResidueSpan として取得できます。
>>> chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
>>> chain.whole()
<gemmi.ResidueSpan of 456: A - K [1(THR) 2(PRO) 3(GLU) ... 456(HOH)]>
subchain
get_subchain(arg: str) や subchains() を使うと、label_asym_id 単位の ResidueSpan をまとめて取得できます。
>>> chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
>>> for subchain in chain.subchains():
print(subchain)
<gemmi.ResidueSpan of 449: A [1(THR) 2(PRO) 3(GLU) ... 449(LYS)]>
<gemmi.ResidueSpan of 1: C [450(ZN)]>
<gemmi.ResidueSpan of 1: D [451(ZN)]>
<gemmi.ResidueSpan of 1: E [452(MG)]>
<gemmi.ResidueSpan of 1: F [453(PO4)]>
<gemmi.ResidueSpan of 3: K [454(HOH) 455(HOH) 456(HOH)]>
ResidueSpanのメソッド
Subchain ID と Polymer Type の取得
GEMMIにおけるsubchainは、CIFファイル内のlabel_asym_idを指します。
ResidueSpan.subchain_id()を呼び出すと、該当するlabel_asym_idが返されます。
また、ResidueSpan.check_polymer_type()を使用することで、_entity_poly.typeに対応するPolymerTypeクラスが返されます。
chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
polymer = chain.get_polymer()
print('Subchain (label_asym_id):', polymer.subchain_id())
print('Polymer Type:', polymer.check_polymer_type())
Subchain (label_asym_id): A
Polymer Type: PolymerType.PeptideL
GEMMIがサポートするPolymerTypeは以下のとおりです。
| 値 | 数値 |
|---|---|
PeptideL |
1 |
PeptideD |
2 |
Dna |
3 |
Rna |
4 |
DnaRnaHybrid |
5 |
SaccharideD |
6 |
SaccharideL |
7 |
Pna |
8 |
CyclicPseudoPeptide |
9 |
Other |
10 |
Unknown |
0 |
各値は、PolymerType.PeptideL.valueのようにして数値にアクセスできます。
chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
polymer = chain.get_polymer()
polymer_type = polymer.check_polymer_type()
if polymer_type.value == 1:
print('PeptideL', 'value:', polymer_type.value)
elif polymer_type.value == 3:
print('Dna', 'value:', polymer_type.value)
elif polymer_type.value == 4:
print('Rna', 'value:', polymer_type.value)
elif polymer_type.value == 0:
print('Unknown', 'value:', polymer_type.value)
else:
print('Other PolymerType', 'value:', polymer_type.value)
PeptideL value: 1
以下のように文章を追加することで、extract_sequence()とmake_one_letter_sequence()の使い分けが明確になります:
配列の取得
GEMMIでは、ResidueSpanオブジェクトを使用して配列情報を取得できます。
-
extract_sequence(): 残基名を3文字形式(例:'THR'や'GLU')のList[str] -
make_one_letter_sequence(): 残基名を1文字形式(例:'T'や'E')のstr
chain = gemmi.read_structure('1alk.cif.gz')[0]['A']
polymer = chain.get_polymer()
print(polymer.extract_sequence()[:10])
print(polymer.make_one_letter_sequence()[:10])
['THR', 'PRO', 'GLU', 'MET', 'PRO', 'VAL', 'LEU', 'GLU', 'ASN', 'ARG']
TPEMPVLENR
Alternative conformations(Microheterogeneity)への対応
first_conformer()で最初のコンフォーマ以外を無視する
>>> polymer = gemmi.read_structure('7a0l.cif.gz')[0]['A'].get_polymer()
>>> print([res.name for res in polymer][30:40])
['GLU', 'MET', 'THR', 'VAL', 'CYS', 'CSO', 'VAL', 'LEU', 'LEU', 'GLU']
>>> print([res.name for res in polymer.first_conformer()][30:40])
['GLU', 'MET', 'THR', 'VAL', 'CYS', 'VAL', 'LEU', 'LEU', 'GLU', 'GLY']
Residue
続いての階層はResidueです。Residueは Atom クラスを保持しています。
Residue から Atom へのアクセス
基本的な操作はSMCRA構造の基本操作をご確認ください。
Atomには__contains__が定義されているため、in構文を使うことができます。
また、__getitem__(name:str)が定義されていますが、Chain->Residueと同様に、返されるのはAtomではなく、AtomGroupになります。
# __contains__
>>> 'CA' in residue:
True
# __getitem__(name:str)
>>> residue['N'] # name:str
<gemmi.AtomGroup N, sites: 1> # nameの場合同名回避でAtomGroupになる
AtomGroup
ResidueGroupのように、同名回避やaltlocに対する対処で導入されています。
個々のAtomにアクセスするには0-based indexもしくはaltloc_idを指定することでアクセスできます。
同名のPDBから取得した構造で同名の残基が被ることはほとんどないとは思いますが、Cα原子を示す'CA'とカルシウムイオンを示す'CA'など、atom_idが被ることはあり得ます。
>>> residue = gemmi.read_structure('1alk.cif.gz')[0]['A'][0]
>>> residue['CA']
<gemmi.AtomGroup CA, sites: 1>
>>> residue['CA'][0] # 0-based indexでアクセス可能
<gemmi.Atom CA at (66.7, 52.9, 2.2)>
# altloc
>>> residue = gemmi.read_structure('11ba.cif.gz')[0]['A']['16']['GLY']
>>> residue['CA']
<gemmi.AtomGroup CA, sites: 2>
>>> residue['CA'].name() # 原子名を取得
'CA'
>>> for atom in residue['CA']:
print(atom.altloc, end=' ')
A B
# altlocで指定のaltlocのAtomにアクセス可能 __getitem__(altloc: str)
>>> residue['CA']['A'] # altloc='A'
<gemmi.Atom CA.A at (4.9, 2.4, 23.9)>
>>> residue['CA']['B'] # altloc='B'
<gemmi.Atom CA.B at (5.4, 2.7, 24.0)>
# 同名(ほとんどの場合ない)
>>> residue = gemmi.read_structure('1alk.cif.gz')[0]['A'][0]
>>> print(residue['CA'])
<gemmi.AtomGroup CA, sites: 1>
# 無理やり追加して検証
>>> atom = gemmi.Atom()
>>> atom.name = 'CA' # 原子名をCA
>>> atom.element = gemmi.Element('Ca') # 元素をCa
>>> print('atom id:', atom.name, 'element:', atom.element)
atom id: CA element: gemmi.Element('Ca')
>>> residue.add_atom(atom) # residueにatomを追加
>>> print(residue['CA']) # AtomGroup, sites: 2
<gemmi.AtomGroup CA, sites: 2>
# この場合、元素名等で判別する必要がある
>>> for i, atom in enumerate(residue['CA']):
if atom.element == gemmi.Element('Ca'):
print('idex:', i, 'atom:', atom)
elif atom.element == gemmi.Element('C'):
print('idex:', i, 'atom:', atom)
else:
print('idex:', i, 'atom:', atom)
idex: 0 atom: <gemmi.Atom CA at (66.7, 52.9, 2.2)>
idex: 1 atom: <gemmi.Atom CA at (0.0, 0.0, 0.0)>
メソッドによるAtomへのアクセス
ResidueにはAtomへアクセスするメソッドがいくつかあります。
find_atom()
find_atom()では残基から特定の原子を取得できます。引数nameで原子名、altlocでaltlocを指定できます。
altlocがある11baの16(GLY)を見てみましょう。
>>> residue = gemmi.read_structure('11ba.cif.gz')[0]['A']['16']['GLY']
>>> list(residue) # altlocの確認
[<gemmi.Atom N.A at (5.8, 1.5, 23.2)>,
<gemmi.Atom N.B at (6.1, 1.7, 23.2)>,
<gemmi.Atom CA.A at (4.9, 2.4, 23.9)>,
<gemmi.Atom CA.B at (5.4, 2.7, 24.0)>,
<gemmi.Atom C.A at (4.2, 1.8, 25.1)>,
<gemmi.Atom C.B at (4.0, 2.8, 23.5)>,
<gemmi.Atom O.A at (4.2, 0.5, 25.2)>,
<gemmi.Atom O.B at (3.3, 3.9, 23.6)>]
# altloc = '*' なら altloc を問わず最初に見つかった原子を返す
>>> residue.find_atom('CA', altloc='*')
<gemmi.Atom CA.A at (4.9, 2.4, 23.9)>
# altloc = 'A' なら altloc = 'A' の原子を探す
>>> residue.find_atom('CA', altloc='A')
<gemmi.Atom CA.A at (4.9, 2.4, 23.9)>
>>> residue.find_atom('CA', altloc='B')
<gemmi.Atom CA.B at (5.4, 2.7, 24.0)>
# altloc = '\0' なら altloc 無しの原子だけ検索
>>> residue.find_atom('CA', altloc='\0')
None # 今回はaltlocしかないのでNone
elでは元素を指定できます(デフォルトはElement('X')で特に指定しない)。
例えば、同じ"CA"でもカルシウムのCAとなっている可能性があり、そのような場合に便利です。
実際にカルシウムイオンがある158dで確認してみましょう
>>> residue = gemmi.read_structure('158d.cif.gz')[0]['A'].get_subchain('C')[0]
>>> print(residue) # カルシウムイオン残基
>>> print(residue[0].element) # カルシウムであることを確認
21(CA)
gemmi.Element('Ca')
# elを指定して検索
>>> print(residue.find_atom('CA', '*')) # el指定なし
<gemmi.Atom CA at (11.2, 7.5, 17.3)>
>>> print(residue.find_atom('CA', '*', gemmi.Element('C')))# 炭素(C)に指定
None
>>> print(residue.find_atom('CA', '*', gemmi.Element('Ca')))# カルシウム(Ca)に指定
<gemmi.Atom CA at (11.2, 7.5, 17.3)>
first_conformer()
最初の簡易的な確認として、first_conformer()を使って最初のconformerのAtomだけを取り出すこともできます。
>>> residue = gemmi.read_structure('11ba.cif.gz')[0]['A']['16']['GLY']
>>> print([atom.name + atom.altloc for atom in residue])
>>> print([atom.name + atom.altloc for atom in residue.first_conformer()])
['NA', 'NB', 'CAA', 'CAB', 'CA', 'CB', 'OA', 'OB']
['NA', 'CAA', 'CA', 'OA']
get_ca() get_p()
get_ca() get_p()で残基からアミノ酸残基ならCα原子、塩基残基ならP原子を取得できます(核酸の例: 101d)。
>>> residue = gemmi.read_structure('1alk.cif.gz')[0]['A'][0]
>>> residue.get_ca()
<gemmi.Atom CA at (66.7, 52.9, 2.2)>
>>> residue = gemmi.read_structure('101D.cif.gz')[0]['A'][1]
>>> residue.get_p()
<gemmi.Atom P at (22.9, 32.8, 20.6)>
Residue に Atom を 追加/削除 する
基本的な操作はSMCRA構造の基本操作をご確認ください。
remove_atom()メソッドを使うとfind_atom()と同じ形で原子を削除することができます。
>>> residue = gemmi.read_structure('11ba.cif.gz')[0]['A']['16']['GLY']
>>> print(residue['CA'])
>>> print(residue.find_atom('CA', 'A'))
<gemmi.AtomGroup CA, sites: 2>
<gemmi.Atom CA.A at (4.9, 2.4, 23.9)>
>>> residue.remove_atom('CA', 'A') # もちろん el: gemmi.Element も指定可能
>>> print(residue['CA'])
>>> print(residue.find_atom('CA', 'A'))
<gemmi.AtomGroup CA, sites: 1>
None
Properties
Residueには多くのプロパティが備わっています。
| Property | データ型 | 備考 |
|---|---|---|
| name | str |
残基名(例: "ALA") |
| seqid.num | int |
シーケンス番号 |
| seqid.icode |
str(長さ1 または空) |
insertion code(例: ' ' または 'A' 等。 ' ' は「なし」を意味) |
| segment | str |
PDB v2 形式の segment |
| subchain | str |
mmCIF の label_asym_id や Structure.assign_subchains() で割り当てられた ID |
| entity_id | str |
mmCIF の _entity.id に対応する文字列 |
| label_seq | int | None |
_atom_site.label_seq_id 等から取得される整数(無い場合は None) |
| entity_type | gemmi.EntityType |
EntityType.Unknown, Polymer, NonPolymer, Water のいずれか |
| het_flag |
str(長さ1 または空) |
'A'=ATOM, 'H'=HETATM, '\0'=unspecified などを表すフラグ |
| flag |
str(長さ1 または空) |
ユーザーが任意に使える追加フラグ |
| sifts_unp | tuple |
SIFTS 注釈などの情報が格納される場合があるタプル |
residue = gemmi.read_structure('1alk.cif.gz')[0]['A'][0]
print('label_asym_id:',residue.subchain)
print('_entity.id:',residue.entity_id)
print('label_seq_id',residue.label_seq)
print('_entity.type:',residue.entity_type)
print('HETATM flag:',residue.het_flag) # 'A'=ATOM, 'H'=HETATM, '\0'=unspecified
label_asym_id: A
_entity.id: 1
label_seq_id 1
_entity.type: EntityType.Polymer
HETATM flag: A
SeqId
seqidはgemmi.SeqIdが返されます。seqid.numが残基番号(_atom_site.auth_seq_id) seqid.icodeがinsertion code(_atom_site.pdbx_PDB_ins_code)に対応します
>>> residue = st = gemmi.read_structure('1igy.cif')[0]['B']['82B'][0]
>>> residue.seqid
<gemmi.SeqId 82B>
>>> print(residue.seqid.num, '+', residue.seqid.icode)
82 + B
# setterの場合もgemmi.SeqIdを使用する
>>> residue.seqid = gemmi.SeqId(999, 'Z') # __init__(番号:int, icode:str)
>>> residue.seqid
<gemmi.SeqId 999Z>
>>> residue.seqid = gemmi.SeqId(999, ' ') # icode必要ないなら' '
>>> residue.seqid
<gemmi.SeqId 999>
Methods
| メソッド名 | 引数 | 返り値の型 | 説明 |
|---|---|---|---|
find_atom |
name: str, altloc: str, el: Element = ... |
Atom |
指定された名前、代替構造、元素に一致する原子を検索します。 |
remove_atom |
name: str, altloc: str, el: Element = ... |
None |
指定された名前、代替構造、元素に一致する原子を削除します。 |
add_atom |
atom: Atom, pos: int = -1 |
Atom |
残基に原子を追加します。 |
first_conformer |
- | FirstConformerAtoms |
最初のコンフォーマを返します(代替構造を無視します)。 |
sole_atom |
arg: str |
Atom |
指定された名前の唯一の原子を返します(一意でない場合は例外をスロー)。 |
get_ca |
- | Atom |
残基のカルボンα原子を取得します。 |
get_p |
- | Atom |
残基のリン原子を取得します(。 |
is_water |
- | bool |
水分子かどうかを判定します。残基名が'HOH','WAT','H2O','DOD'の場合Trueを返します。'OH'や'H3O'を含めたその他の残基はFalseを返します。 |
remove_hydrogens |
- | None |
残基から水素原子を削除します。 |
trim_to_alanine |
- | bool |
残基をアラニンにトリムします。 |
recommended_het_flag |
- | str |
推奨されるHETATMフラグを返します。 |
clone |
- | Residue |
残基をコピーします。 |
ほとんど上記で紹介したので説明は割愛します。
trim_to_alanine()はなぜかうまく機能しません (ver. 0.7.0)
Atom
最後に、最下層のAtomになります。最下層ということもあり、下層へのアクセスや追加削除といったことはできません。
Atomのプロパティ
| プロパティ | 型 | 説明 |
|---|---|---|
| name | str |
原子名 (例: "CA", "CB" など)。補足: PDBフォーマットで桁合わせする場合は atom.padded_name() を利用可能。 |
| altloc |
str(長さ1または空) |
オルタナティブロケーション (例: 'A', 'B', '\0'=なし)。atom.has_altloc() で非ヌルかどうかを判定できる。 |
| charge | int |
整数値の電荷(部分電荷はサポートされない)。 |
| element |
Element (gemmi.Element) |
元素(周期表)。atom.is_hydrogen() で水素(重水素を含む)かどうかを判定できる。 |
| pos | Position | Sequence[float] |
座標(Å単位)。atom.pos = [x, y, z] のようにリストでも設定可能。 |
| occ | float |
占有率。 |
| b_iso | float |
等方性 B因子(原子変位パラメータ)。 非等方性 ADP ( aniso) と併せて扱う場合がある。 |
| aniso |
SMat33f (gemmi.Anisotropy 相当) |
非等方性原子変位パラメータ(Uパラメータ、6成分)。atom.aniso.nonzero() で無設定かどうかを確認可能。 |
| serial | int |
原子シリアル番号。 |
| fraction | float |
mmCIF の _atom_site.occupancy や、一部構造解析で使用される場合がある補助的パラメータ。 |
| calc_flag |
CalcFlag (gemmi.CalcFlag) |
mmCIF の _atom_site.calc_flag に対応。2020年以降の mmCIF 仕様で追加された計算由来フラグなどに用いられることがある。 |
| flag |
str(長さ1または空) |
ユーザー任意のフラグ。 |
| tls_group_id | int |
TLS (Translation-Libration-Screw) グループID。 結晶学的解析などで各原子を複数グループに分ける際に用いられる。 |
atom = gemmi.read_structure('11ba.cif.gz')[0]['A']['16']['GLY']['CA'][0]
print(atom.name) # _atom_site.label_atom_id
print(atom.altloc) # _atom_site.label_alt_id
print(atom.element) # gemmi.Element('C'), _atom_site.type_symbol
print(atom.pos) # <gemmi.Position(x, y, z)>, _atom_site.Cartn_x/y/z
print(atom.occ) # _atom_site.occupancy
print(atom.b_iso) # _atom_site.B_iso_or_equiv
print(atom.serial) # _atom_site.id
CA
A
gemmi.Element('C')
<gemmi.Position(4.936, 2.41, 23.894)>
0.5
27.020000457763672
123
Atomのメソッド
| メソッド名 | 引数 | 返り値の型 | 説明 |
|---|---|---|---|
is_hydrogen |
- | bool |
この原子が水素原子かどうかを判定します。 |
has_altloc |
- | bool |
この原子に代替構造(altloc)が設定されているかを判定します。 |
b_eq |
- | float |
異方性B因子を等方性B因子に変換した値を計算して返します。 |
padded_name |
- | str |
原子名をPDB形式のように左詰めされた名前として返します。 |
clone |
- | Atom |
この原子のコピーを返します。 |
Position
Atom.posに収録されるgemmi.Positionでは原子のxyz座標(ベクトル)を取り扱えます。Python の数値演算と同様の使い方ができるほか、dist()で2座標間のユークリッド距離が簡単に求められます。また、スカラ積や除算のほか、符号反転(-pos)などもサポートされ、直感的なベクトル操作を行えます。
プロパティ(オーバーライド元のVec3のも含む)
| プロパティ | 型 | 説明 |
|---|---|---|
| x | float | x 座標 |
| y | float | y 座標 |
| z | float | z 座標 |
メソッド (オーバーライド元のVec3のも含む)
| メソッド名 | 引数 | 返り値の型 | 説明 |
|---|---|---|---|
normalized |
self |
Vec3 |
自身を正規化(長さを1に)した新たなVec3を返す(元のベクトルは変更しない)。 |
dot |
self, arg: Vec3 |
float |
自身と別のVec3の内積を返す。 |
cross |
self, arg: Vec3 |
Vec3 |
自身と別のVec3の外積を返す。 |
length |
self |
float |
ベクトルの長さ(ユークリッドノルム)を返す。 |
approx |
self, other: Vec3, epsilon: float |
bool |
自身がotherと近い(各成分の差がepsilon以下)かどうかを返す。 |
tolist |
self |
list[float] |
(x, y, z)をリスト[x, y, z]に変換して返す。 |
fromlist |
self, arg: Sequence[float] |
None |
[x, y, z]形式の配列やリストから値を読み込み、自身を上書きする。 |
dist |
self, arg: Position |
float |
自身と別のPositionの間のユークリッド距離を返す(√((x2-x1)² + (y2-y1)² + (z2-z1)²))。 |
pos = gemmi.read_structure('1alk.cif.gz')[0]['A'][0]['CA'][0].pos
print(pos) # Position
print(pos.x, pos.y, pos.z) # x, y, z
print(pos.length()) # ユーグリットノルム
print(pos.normalized()) # 正規化Vec
print(pos.tolist()) # list[x, y, z]
<gemmi.Position(66.663, 52.938, 2.184)>
66.663 52.938 2.184
85.15372727602708
<gemmi.Vec3(0.782855, 0.621676, 0.0256477)>
[66.663, 52.938, 2.184]
pos_2 = gemmi.read_structure('1alk.cif.gz')[0]['A'][1]['CA'][0].pos
print(pos.dist(pos_2)) # ユーグリット距離
print(pos.approx(pos_2, 5.0)) # 5.0以内にあるか
print(pos.dot(pos_2)) # 内積
print(pos.cross(pos_2)) # 外積
3.8033532573243867
True
7191.1795170000005
<gemmi.Vec3(127.498, -150.243, -249.931)>
print(pos + pos_2)
print(pos - pos_2)
print(pos * 2)
print(pos / 2)
print(-pos)
<gemmi.Position(134.554, 103.102, 6.662)>
<gemmi.Position(-1.228, 2.774, -2.294)>
<gemmi.Position(133.326, 105.876, 4.368)>
<gemmi.Position(33.3315, 26.469, 1.092)>
<gemmi.Position(-66.663, -52.938, -2.184)>
等方性 (Isotropic) と 非等方性 (Anisotropic) ADP
ChatGPTに説明させたものをそのまま載せています
b_iso
- 等方性 B 因子を表すプロパティ。
- 数式的には ( $B \approx 8\pi^2 U_{\text{iso}}$ ) の関係があり、PDB・mmCIF などでは「B 因子(B-factor)」として表記される。
aniso (atom.aniso)
- 非等方性 ADP(U パラメータ:( $U_{11}, U_{22}, U_{33}, U_{12}, U_{13}, U_{23}$ ))を格納。
-
atom.aniso.nonzero()によって非等方性パラメータが有効か(すべて 0 ではないか)を確認できる。
PDB 形式
- 等方性 ADP は
Bとして、非等方性はUとして保存。($B = 8 \pi^2 U$)
-
b_eq()とcalculate_b_est()(Gemmi での計算)-
b_eq()-
非等方性 ADP から 等価等方性 B 因子 (B_eq) を計算する関数。
-
定義:
B_\mathrm{eq} = 8\pi^2 \cdot \frac{(U_{11} + U_{22} + U_{33})}{3} -
非等方性パラメータの平均的な動きを、等方性で近似した B-factor として取得する。
-
-
gemmi.calculate_b_est(atom)-
B_est を計算する関数。Ethan Merrit が提案した指標で、
b_eq()よりも実際の等方性精密化で出る B に近い値を得られるように定義されたもの。 - PDB データによっては “residual” B 因子が格納されている場合があり、その場合に B_iso などとの比較に使うと有用。
-
B_est を計算する関数。Ethan Merrit が提案した指標で、
-
-
PDB データの注意点
- PDB/MMCIF では、非等方性モデルでも等方性 B 因子に相当する値(Biso や B_iso)を同時に保存している場合がある。
- ただし、一部のエントリでは “residual” B 因子を
B_isoに詰めている例もあり、厳密に「フル等方性 B 因子」ではないケースがある点に注意。 -
b_eq()やb_estを計算することで、“非等方性パラメータから換算された等方性 B” をユーザが自由に評価できる。
終わりに
GEMMIを使ったタンパク質立体構造解析の基礎(SMCRAデータ構造)をまとめてみました。GEMMIはCIFファイル形式に特化しており、BioPythonでは曖昧であったaltlocなどの扱いがより厳密になっています。
今回はSMCRAデータ構造のみでしたが、SelectionやSuperposeといった発展的な解析も行えるので、次回はそちらもまとめてみたいと思っています。