さいしょに
この記事は、RustBCAのtomlファイルについての公開メモ帳みたいなものです。こんな感じかなあ、というノリで書いているので、内容についてはあまり信用しないでほしいのですが、全く適当という訳ではないです。
RustBCAについては下の記事を読んでください。
input file option
RustBCAは、tomlファイルでシミュレーション条件を設定します。wikiのinputで説明されてるので、Gemini等で翻訳すればある程度は理解できるのですが、それでも適当に触ればハマります。
この記事ではそのハマり点などを随時メモっておく、みたいな形になっています。layered_geometry.tomlをベースに説明するような形式をとります。
設定項目は、[options] [particle_parameters] [geometry_input] [material_parameters] の4つの大項目に分かれています。それぞれ見ていきましょう。
[options]
作成中
[particle_parameters]
[particle_parameters] は入射する粒子(イオン)のパラメーターを定義します。
他のイオン種でシミュレーションしたい場合はこのあたりを変えるべきでしょうか。多価イオンの場合はどうするのかな。
N = [ 10000,] #シミュレーションする粒子の数
m = [ 4.002602,] #粒子の質量 (mass_unitでAMUを定義していれば原子質量単位)
Z = [ 2,] #原子番号(He)
E = [ 2000.0,] #初期エネルギー(energy_unitでEVを定義していれば、イオン注入における加速電圧と考えて良いと思う)
入射角度とかを変えたい場合はこのあたりを変えるのだと思います。
pos = [ [ -1.7453292519934434e-8, 0.0, 0.0,],]
dir = [ [ 0.999, 0.001, 0.0,],]
posがこの値になっている理由はわかりませんが、Geminiによると0だと浮動小数点の揺らぎで物質内部から開始してしまうことも起こりえるため、0を設定せずほんの少し表面側を指定するとのことです。このシミュレーションは、x軸の左(マイナス)から右(プラス)へ向かって粒子を飛ばす、という設定になっています。ターゲット表面の座標は[geometry_input]で定義しますが、x=0にしておくのがわかりやすくて良いでしょう。なので、-1.7453...e-8は多分-2.0e-8とかにしても問題ないので、シンプルな数値にしておいた方が後の人に親切な気がします。
dirは、入射イオンの初期進行方向をベクトル(x, y, z成分)のリストで指定します。ターゲットが単結晶の場合、チャネリングを防ぐため少し傾きを持たせることがあるときは、ここで設定します。
[geometry_input]
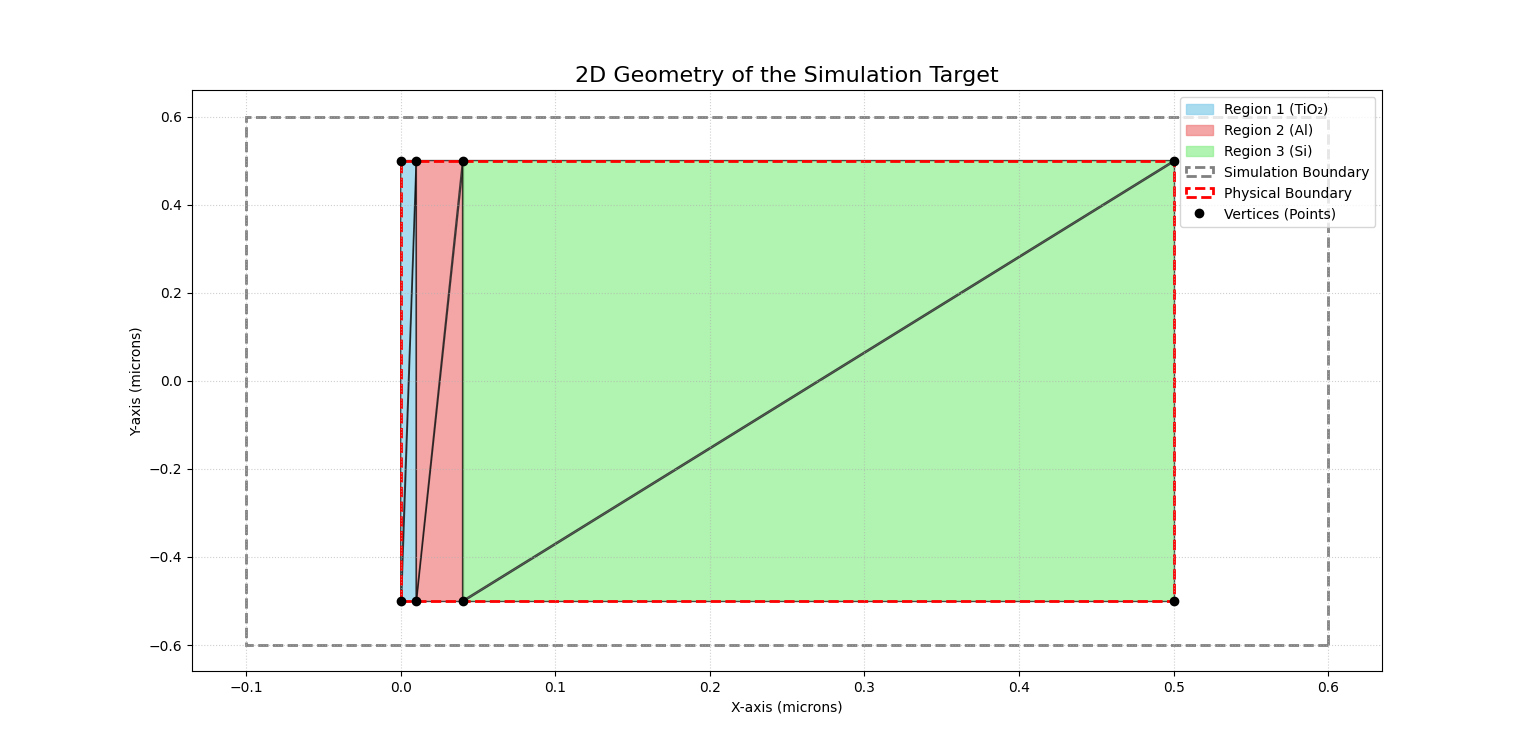
[geometry_input] はターゲット側の形状と組成を定義します(物理的特性はここでは定義しない)。points、triangles、densitiesが最も重要な設定かと思います。
それぞれのパラメーターの数(listサイズ)が合っていないと、シミュレーションが開始せずエラーを返します。
-
length_unitは長さ(距離)の単位を設定します。layered_geometry.tomlでは"MICRON"が設定されています。ANGSTROM, NM, MICRON, CM, MM, M,もしくは"1e-6"(メートル)という形での設定が可能です。ここをミスると全てが破綻します。 -
pointは メッシュを構成する頂点を設定します。layered_geometry.tomlでの設定をわかりやすく書くと下のようになります。
points = [
[0.0, -0.5], # 頂点0
[0.01, -0.5], # 頂点1
[0.04, -0.5], # 頂点2
[0.5, -0.5], # 頂点3
[0.5, 0.5], # 頂点4
[0.04, 0.5], # 頂点5
[0.01, 0.5], # 頂点6
[0.0, 0.5] # 頂点7
]
つまり、x=0, y=-0.5の頂点を0番目の頂点、x=0.01, y=-0.5の頂点を1番目としているわけです。ここで、xは0, 0.01, 0.04, 0.5で区切っているのに対し、yは-0.5と0.5しかないことに気付くかと思います。これは、xが深さを示し、二酸化チタン(TiO₂)/ アルミニウム(Al)/ シリコン(Si)の3層を設定するために3分割(0と0.5は外部との境界)しており、yは一様なので区切ってないわけです。(この場合、TiO2が10nm, Alが30nm、Siが残り500nmまで、多分Si基板上に製膜したサンプルを想定しているのでしょう)
-
trianglesは各三角形を構成する点を指定するリストです。layered_geometry.tomlでの設定をわかりやすく書くと下のようになります。
triangles = [
[0, 1, 6], #三角形0 頂点0と1と6を繋ぐ三角形
[0, 6, 7], #三角形1 頂点0と6と7を繋ぐ三角形
[1, 2, 5], #三角形2 頂点1と2と5を繋ぐ三角形
[1, 5, 6], #三角形3 頂点1と5と6を繋ぐ三角形
[2, 3, 4], #三角形4 頂点2と3と4を繋ぐ三角形
[2, 4, 5] #三角形5 頂点2と4と5を繋ぐ三角形
]
つまり、三角形0の頂点は[0.0, -0.5],[0.01, -0.5],[0.01, 0.5]をつないだものになります。x=0近傍の超細長い三角形で、サンプルの表面近傍の領域を三角形で示します。
-
densitiesは各三角形領域に含まれる原子の密度を設定しています。layered_geometry.tomlでの設定をわかりやすく書くと下のようになります。
densities = [
[ 3.0e+10, 6.0e+10, 0.0, 0.0,], #三角形0の密度 マテリアル0の密度が3.0e+10 atoms/µm3, マテリアル1の密度が6.0e+10 atoms/µm3
[ 3.0e+10, 6.0e+10, 0.0, 0.0,], #三角形1の密度 たいへんなので略
[ 0.0, 0.0, 6.026e+10, 0.0,],
[ 0.0, 0.0, 6.026e+10, 0.0,],
[ 0.0, 0.0, 0.0, 4.996e+10,],
[ 0.0, 0.0, 0.0, 4.996e+10,],
]
ここでは4つのマテリアルの密度を設定します。物理特性は、後で設定するmaterial_parametersにて定義します。
三角形0と1は同じ密度が設定されており、ここでは1:2だなあという事しかわからないかもしれませんが、material_parametersのほうを見ると、Ti(マテリアル0)とO(マテリアル1)の物質特性が入力されている事がわかります。ちなみにマテリアル2はAl, マテリアル3はSiです。つまり、表面から見て1層目はTiO2, 2層目がAl, 3層目(残り、基板)がSiの密度に対応しています。
最初にlength_unit = "MICRON"としているので、ここの単位はatoms/µm3です。ここをミスってもシミュレーション自体はエラーを吐かないのですが、変な結果を返すことになります。
(注入したのがどっか行っちゃったな~とか何回かやった)
-
boundaryはターゲットの外との境界を定義します。pointsを見れば、この点が一番外側の四隅に対応している事がわかります。
boundary = [0, 3, 4, 7] #頂点0と3と4と7が境界
-
simulation_boundary_pointsはシミュレーションの境界を定義します。この枠から出たらシミュレーションを打ち切ることになります。pointsの少し外側とし、最初の点と最後の点が同じになるように閉じた領域を設定する必要があります。
simulation_boundary_points = [ [ 0.6, -0.6,], [ -0.1, -0.6,], [ -0.1, 0.6,], [ 0.6, 0.6,], [ 0.6, -0.6,],]
まとめると、下の図のようにターゲット側を定義しています。
[material_parameters]
ターゲット材料の物理的特性を定義します。layered_geometry.tomlでは下のように書かれています。
energy_unit = "EV"
mass_unit = "AMU"
Eb = [ 3.0, 2.58, 3.0, 0.0,]
Es = [ 4.84, 2.58, 3.39, 4.72,]
Ec = [ 3.5, 2.0, 3.0, 1.5,]
Z = [ 22, 8, 13, 14,]
m = [ 47.867, 15.9994, 26.98, 28.08553,]
interaction_index = [0, 0, 0, 0]
surface_binding_model = {"PLANAR"={calculation="TARGET"}}
bulk_binding_model = "AVERAGE"
energy_unitとmass_unitは他の項目と同じように設定しておけは良いでしょう。
EbはBulk binding energy、EsはSurface binding energy、EcはCutoff energy、Zは原子番号、mは原子の質量です。geometry_inputで4つの原子(Ti, O, Al, Si)を定義したので、ここでもリストで4つの物理特性を定義します。
基本的にEbとEsは文献を調べて指定する必要があります。しかし、この設定で不思議なのはSiのEbに0が指定されているところです。おそらく、TiO2とAlの膜でシミュレーションをしたくて、Siはまあ基板なのでまじめに計算しなくても良い、という気もちで0を入れたのだと思われます。
Ecはこれ以下のエネルギーになったら計算を止める、という閾値なので、目的に合わせて指定する必要があります。基本的に小さい値のほうが、「注入した原子がどこまでとどくか」みたいなシミュレーションをしたい場合に有効でしょう。大きい値のほうが計算が短縮されるため、Ebと同じか、ちょっと上かちょっと下あたりを設定するのがちょうど良いかもしれません。
surface_binding_modelとbulk_binding_modelはよく理解していません。ちゃんと教科書を読まないといけないですが、とりあえずデフォルトで良いんじゃないかな...
シミュレーション例
BをSiに注入
[options]
# シミュレーション名:BをSiに注入
name = "boron_on_si"
# 粒子の軌跡は追跡しない
track_trajectories = false
track_recoils = true
track_recoil_trajectories = false
write_buffer_size = 8000
weak_collision_order = 3
suppress_deep_recoils = false
high_energy_free_flight_paths = false
electronic_stopping_mode = "LOW_ENERGY_NONLOCAL"
mean_free_path_model = "LIQUID"
interaction_potential = [["KR_C"]]
scattering_integral = [["MENDENHALL_WELLER"]]
use_hdf5 = false
root_finder = [[{"NEWTON"={max_iterations=100, tolerance=1E-6}}]]
num_threads = 4
num_chunks = 10
track_displacements = false
track_energy_losses = false
energy_min = 0.0
energy_max = 50000.0
energy_num = 20
angle_min = 0.0
angle_max = 90.0
angle_num = 20
x_min = 0.0
y_min = -0.1
z_min = -0.1
x_max = 1.0
y_max = 0.1
z_max = 0.1
x_num = 20
y_num = 20
z_num = 20
[particle_parameters]
length_unit = "1e-6"
energy_unit = "EV"
mass_unit = "AMU"
N = [10000,]
m = [10.811,]
Z = [5,]
E = [35000.0,]
Ec = [1.0,]
Es = [0.0,]
interaction_index = [0]
pos = [ [ -2E-8, 0.0, 0.0,],]
dir = [ [ 0.999, 0.001, 0.0,],]
[geometry_input]
length_unit = "MICRON"
# Si単一基板の形状を定義(単純な矩形)
points = [[0.0, -1.0], [1.0, -1.0], [1.0, 1.0], [0.0, 1.0]]
triangles = [[0, 1, 2], [0, 2, 3],]
# Siの原子密度: 約5.0e+22 atoms/cm^3
densities = [[5.0e+10,], [5.0e+10,],]
boundary = [0, 1, 2, 3]
simulation_boundary_points = [ [ 1.1, -1.1,], [ -0.1, -1.1,], [ -0.1, 1.1,], [ 1.1, 1.1,], [ 1.1, -1.1,],]
electronic_stopping_correction_factors = [1.0, 1.0]
energy_barrier_thickness = 2.2E-4
[material_parameters]
energy_unit = "EV"
mass_unit = "AMU"
# Siの結合エネルギーなど
Eb = [4.72,]
Es = [4.72,]
Ec = [1.0,]
# Siの原子番号と質量
Z = [14,]
m = [28.08553,]
interaction_index = [0,]
surface_binding_model = {"PLANAR"={calculation="TARGET"}}
bulk_binding_model = "AVERAGE"
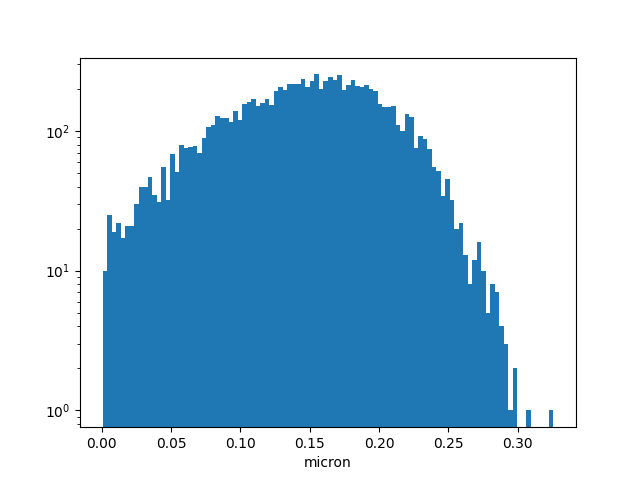
と比較してもピーク位置ちょっとずれてるナア、これがどの程度のものなのか...裾野も後処理か何かで調整必要だなあ