SDFの読み込みにおいて、ありがちなトラブルにを紹介し、解決方法も解説します。なお、この記事は創薬 Advent Calendar 2017 (#souyakuAC2017) の19日目の記事です。
構造式が潰れてしまう
次のプログラムは、3日目の記事でPubChemから取得したyes1_inhibition.sdfに収録された化合物のうち、最初の10個について構造式を描画しようとしたプログラムです。
from rdkit import Chem
from rdkit.Chem import Draw
spl = Chem.SDMolSupplier('yes1_inhibition.sdf')
mols = []
for mol in spl:
mols.append(mol)
# the first 10 compounds are extracted.
mols = mols[:10]
img = Draw.MolsToGridImage(mols, molsPerRow=5,subImgSize=(300, 300))
img.save("molecules.png")
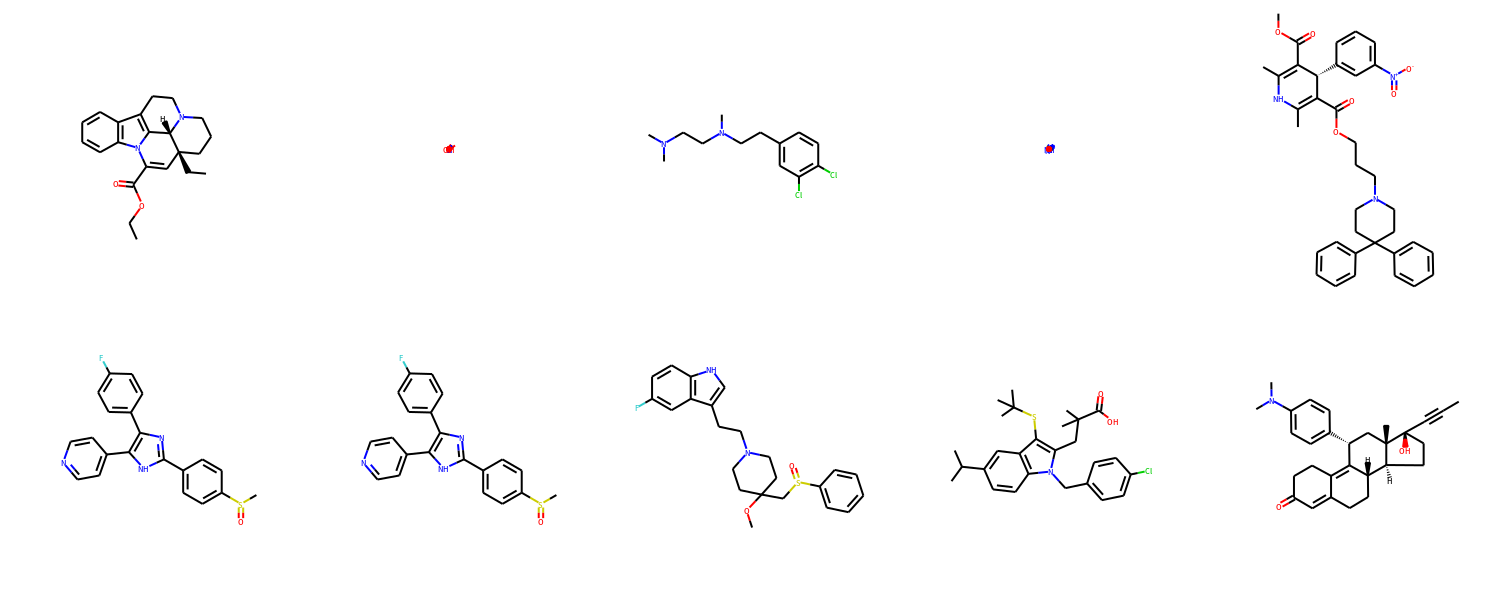
2番目と4番目の化合物が凝集したように潰れてしまって、きちんと描画できていません。yes1_inhibition.sdfをテキストエディタかlessコマンドで開いて、原因を探りましょう。まずは問題なく描画できている1番目の化合物、つまりファイルの先頭部分を確認してみます。
11112768
-OEChem-11231705332D
26 30 0 1 0 0 0 0 0999 V2000
-0.6502 -4.8379 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-2.4607 -3.3498 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.9700 -0.9800 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
1.9800 1.1100 0.0000 N 0 0 3 0 0 0 0 0 0 0 0 0
-0.2000 0.0900 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2000 0.0000 0.0000 C 0 0 3 0 0 0 0 0 0 0 0 0
小数が各行に3個並んでいますね。察しのいい方は気がついたでしょうが、これは化合物を構成する各原子の座標です。
3番目がいずれも0であることから、Z軸の情報は持たない2Dの情報が保持さていることが分かります。
さて、このままSDFの内容を画面スクロールをしていくと、$$$$とだけ記載のある行に到達するはずです。これはある化合物と次の化合物の区切りのサインです。つまり、これ以降に記述されているのが問題の2番目の化合物ということになります。
$$$$
11113545
-OEChem-11231705332D
19 20 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2番目の化合物は、すべての原子の座標が(0, 0, 0)の原点に位置しています。これが構造式が潰れてしまった原因ですね。座標情報が実質的に含まれていない手抜きな化合物が紛れ込んでいたのでした。こういう場合でも原子間の結合の情報がきちんと保持されてれば、2次元の座標情報はRDKitの機能で構築することができます。
from rdkit import Chem
from rdkit.Chem import Draw, AllChem
spl = Chem.SDMolSupplier('yes1_inhibition.sdf')
mols = []
for mol in spl:
# compute 2D coordinate
AllChem.Compute2DCoords(mol)
mols.append(mol)
# the first 10 compounds are extracted.
mols = mols[:10]
img = Draw.MolsToGridImage(mols, molsPerRow=5,subImgSize=(300, 300))
img.save("molecules.png")
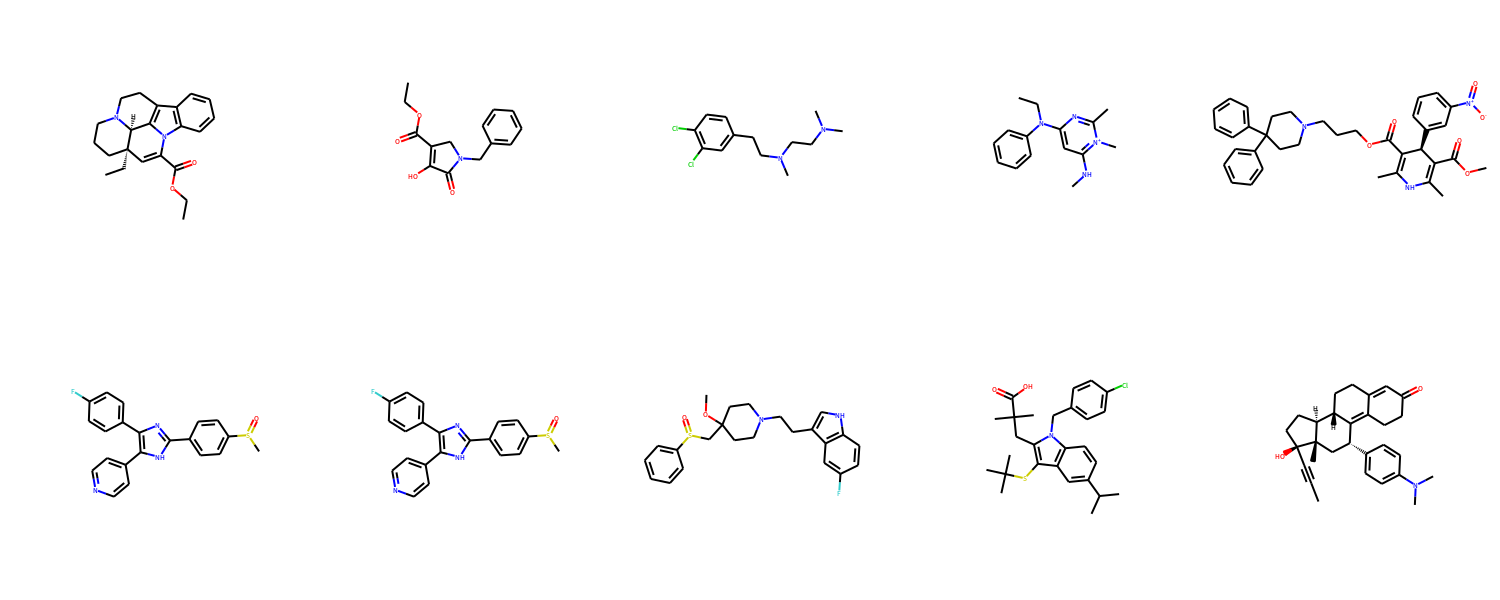
Compute2DCoords関数で2次元の座標を構築することで、すべての化合物についてまともな構造式が描画できました。
異常な原子価が引き起こす警告とエラー
今度は別のSDFを下記のURLから取得して、さきほど修正したプログラムで読み込んでみて下さい 1。
すると、以下のようなエラーが発生するはずです。
[03:05:23] Explicit valence for atom # 7 N, 4, is greater than permitted
[03:05:23] ERROR: Could not sanitize molecule ending on line 472
[03:05:23] ERROR: Explicit valence for atom # 7 N, 4, is greater than permitted
Traceback (most recent call last):
File "draw_compounds.py", line 10, in <module>
AllChem.Compute2DCoords(mol)
Boost.Python.ArgumentError: Python argument types in
rdkit.Chem.rdDepictor.Compute2DCoords(NoneType)
did not match C++ signature:
Compute2DCoords(RDKit::ROMol {lvalue} mol, bool canonOrient=True, bool clearConfs=True, boost::python::dict {lvalue} coordMap={}, unsigned int nFlipsPerSample=0, unsigned int nSample=0, int sampleSeed=0, bool permuteDeg4Nodes=False, double bondLength=-1.0)
なんだか難解なエラーが数行にわたって出ていますが、根本的原因はExplicit valence for atom # 7 N, 4, is greater than permittedの部分です。Nの原子価が4になっていて、これが許容される原子価よりも大きい、というのがRDKitの言い分です。確かに化学の常識からすれば中性の窒素原子の原子価は3ですから、通常ありえない構造の化合物を読み込もうとして失敗したということは理解できます。また、RDKitは、このようなケースでは、molにNoneをセットする仕様になっています。その結果、期せずしてNoneがCompute2DCoordsに渡されてしまったことが、プログラムがエラーを吐いてしまったことの直接の原因です。
この場合、molにNoneがセットされていたときには、処理をスキップしてエラーを回避するのが手っ取り早い解決策です2。
from rdkit import Chem
from rdkit.Chem import Draw, AllChem
# from https://pubchem.ncbi.nlm.nih.gov/bioassay/624095
spl = Chem.SDMolSupplier('antibiotics.sdf')
mols = []
for mol in spl:
if mol is None:
continue
# compute 2D coordinate
AllChem.Compute2DCoords(mol)
mols.append(mol)
# first 10 compounds are extracted.
mols = mols[:10]
img = Draw.MolsToGridImage(mols, molsPerRow=5,subImgSize=(300, 300))
img.save("molecules.png")
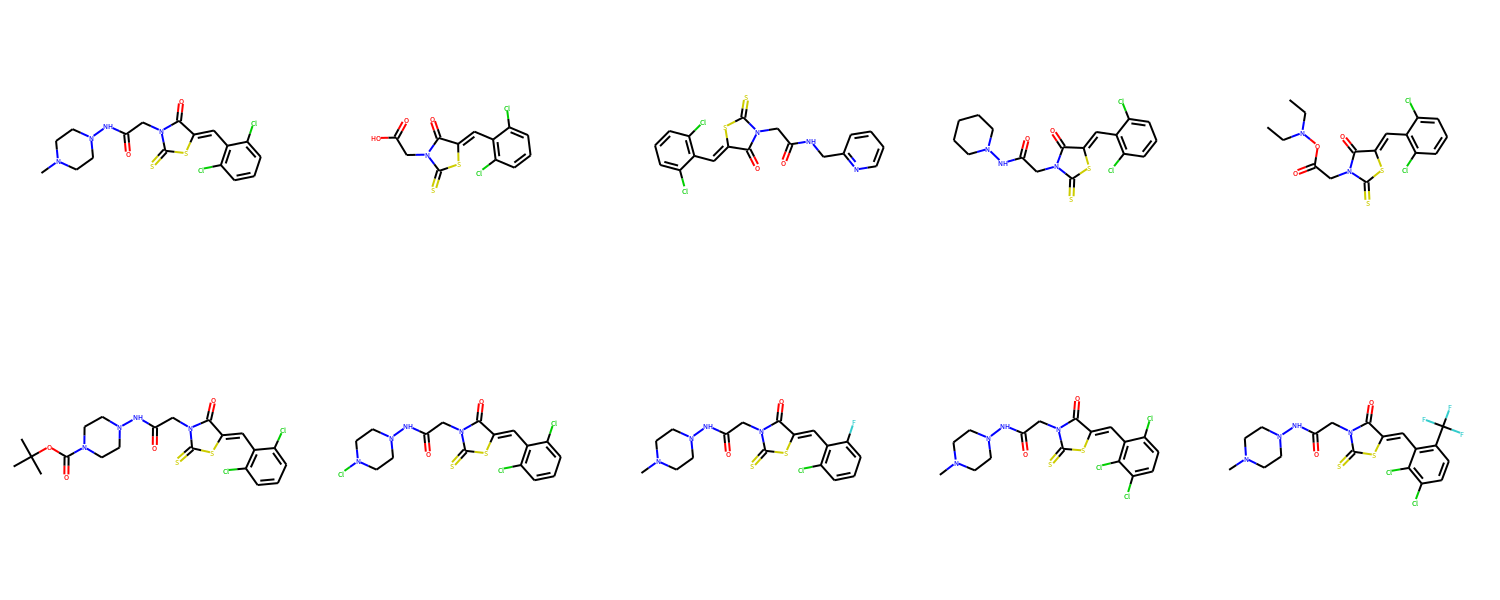
上記のプログラムでも原子価に関する警告は引き続き表示されますが、誤ってNoneがCompute2DCoordsに渡されることはなくなるため、エラーは発生しません。