これから化合物データ処理に取り組む人を対象として、化合物データの代表的なフォーマットとRDKitによる初歩的な取り扱いについて書きます。なお、この記事は創薬 Advent Calendar 2017 (#souyakuAC2017) の3日目の記事です。
化合物データのフォーマット
実際には、様々なフォーマットが使われていますが、最も広く利用されていると思われるSDFとSMILESの2形式について説明します。
SDF
structure-data fileの略。実は、Molfileと呼ばれるフォーマットをベースにして、複数の化合物を1ファイルに格納できるように拡張された形式です。
それ以外の特徴としては、各化合物について任意のプロパティを持つことができることが挙げられます。例えば、化合物のID、分子量、活性値等。
仕様上は化合物中の各原子について3次元の座標を保持できますが、実際にはZ軸の値がすべて0になっていて実質2次元の情報しか保持していない場合も多いです。
フォーマットの中身は下記の記事が分かりやすいです。
SMILES
厳密にはファイル・フォーマットではなく、化合物の表記法。分岐や環構造を含む構造式を文字列に変換したもので、コンピュータでの扱い易さと人間の見やすさを両立しているのが良いところです。例を挙げると、以下のようになります。
| 化合物名 | 構造式 | SMILES文字列 |
|---|---|---|
| エタノール | 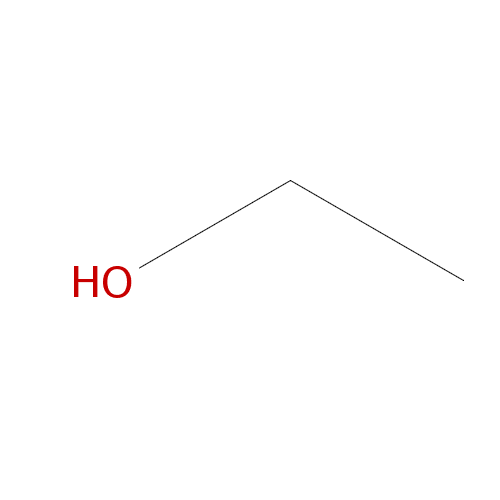 |
CCO |
| 2-プロパノール | 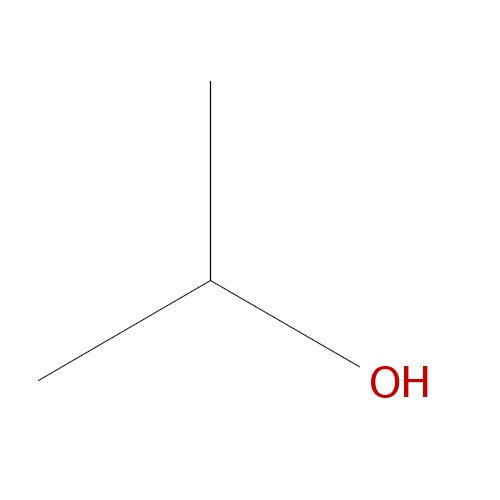 |
CC(C)O |
| アセチルサリチル酸 | 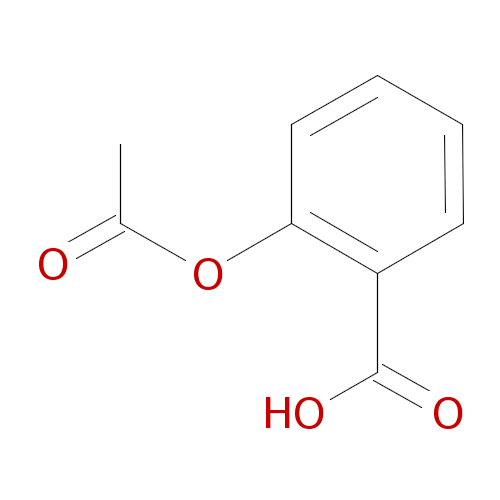 |
CC(=O)Oc1ccccc1C(=O)O |
SMILES文字列を読み解く上でのポイントは下記の通りです。:
- 水素は理由がなければ省略される。
- 分岐は括弧書きで表現される。
- 芳香環中の原子は小文字で表記。
- 単結合は基本省略、二重結合は「=」、三重結合は「#」で表記。
- 分岐後の合流 (つまり、環)は、合流する2原子の後に同一の番号を書いて表現。
ここに列挙したのは一部で、実際にはありとあらゆる構造式が表記できるようにそれなりに複雑なルールが用意されています。正確な仕様についてはOpenSMILES Home Pageを参照しましょう。
ところで、ちょっと考えれば分かることですが、構造式には始点となる原子が明確に存在するわけではありませんし、分岐に突き当たったときにどの鎖を括弧書きにするかは任意なので、単一の化合物を複数の種類のSMILES文字列で表記することができます。例えば、エタノールはヒドロキシル基側から書いて「OCC」と書くこともできて、これも有効なSMILESです。
しかし、これでは例えば2つのSMILES文字列が与えられたときに、それが2種類の異なる化合物を記述しているのか、実は同一の化合物を記述しているのか簡単には判断できません。そこで作為的なルールを追加して、化合物と1対1対応するようにしたSMILESが使われることがあり、この特別なSMILESをcannonical SMILESと呼びます。
SMILES文字列はSDFとは異なり、構造式と等価の情報、つまり2次元の構造情報しか保持することができません。また、先述の通り、ファイル・フォーマットでなく表記法であるため、CSVのテーブルの中で用いるなど柔軟な使い方ができるメリットがあります。ただし、その反面、誰かに「SMILESで化合物データを下さい」と言った場合に送られてくるファイルのフォーマットが、単に一行ずつSMILES文字列を書いたテキストファイルなのか、CSV形式なのか、はたまたエクセルファイルなのかは分かりません。
化合物データの入手元
最も簡単な方法は、ChEMBLやPubChemなどの公共データベースから入手する方法です。
公共データベースの使い方については、下記の統合TVの動画が参考になります。
ここでは、後述のRDKitのプログラムで読み込むために、Yes1キナーゼの阻害実験の対象とされた化合物を下記のページからダウンロードしてみましょう。
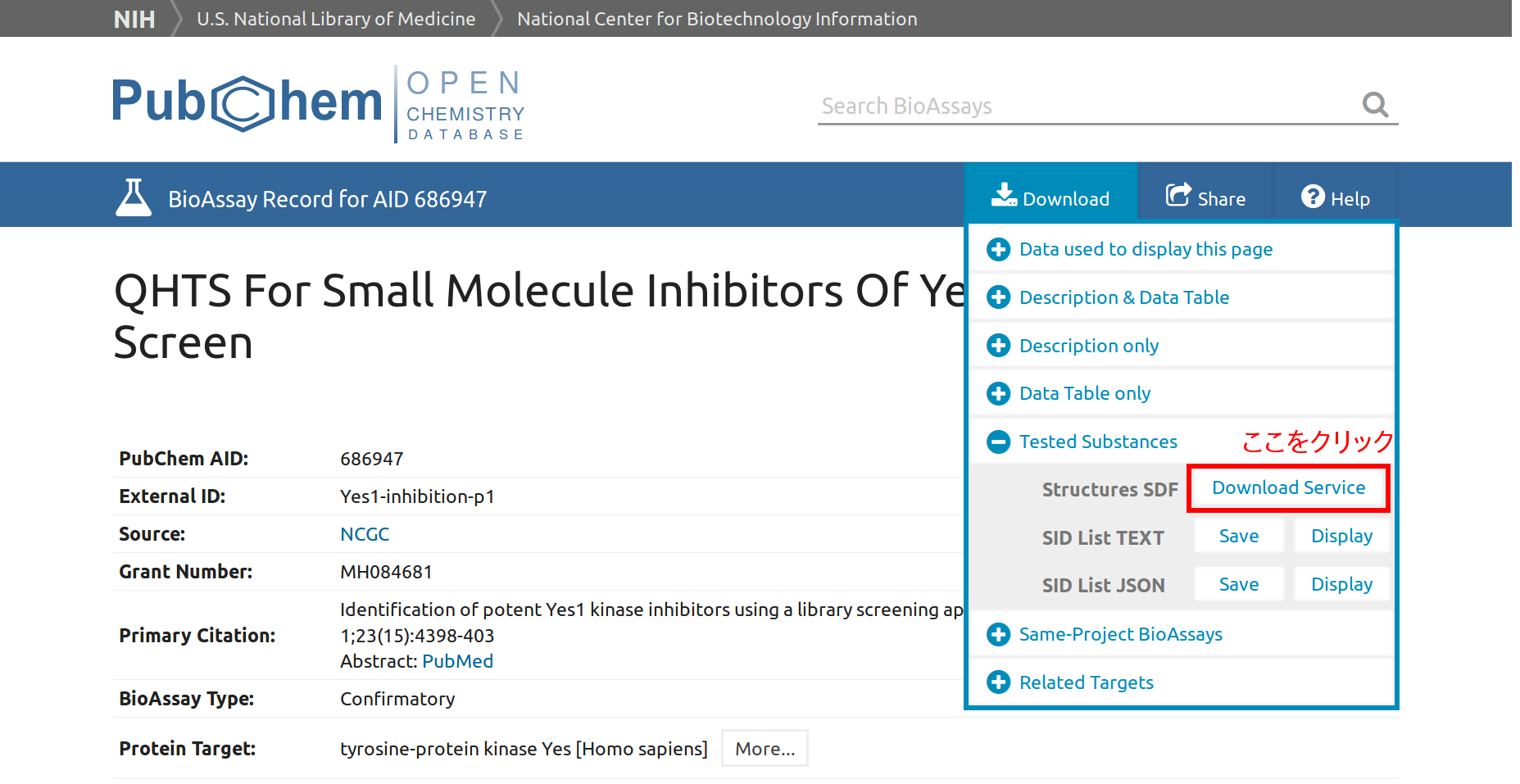
ダウンロードされたファイルのファイル名はPubChemの仕様のために適当な番号になってしまうので、分かりやすいように「yes1_inhibition.sdf」にリネームしておきましょう。
RDKitで化合物データを読み込む
SDFやSMILESなど化合物データを読み込む手っ取り早いのは、RDKitやCDKのようなケモインフォマティクスのためのライブラリを使う方法です。RDKitはPython・C++用、CDKはJava用のライブラリです。ここではRDKitを使って化合物を読み込む練習をしつつ、ついでにごく簡単な分析をやってみましょう。
RDKitのインストール
RDKitはソースコードのコアな部分がC++で書かれ、それがPythonのコードでラップされた作りになっています。ソースコードからビルドすることもできますが、boost関係のエラーでうまく行かないことが多いので、初心者向けとは言い難いです。簡単なのは、公式の案内に従ってコンパイル済みのバイナリをanacondaあるいはminicondaでインストールする方法です。
SDFの読み込みと分子量の計算
先ほどダウンロードしたSDFに記録された各化合物について分子量を計算し、化合物数、最小/最大/平均の分子量を出力するプログラムです。
from rdkit import Chem
from rdkit.Chem import Descriptors
spl = Chem.SDMolSupplier('yes1_inhibition.sdf')
mws = []
for mol in spl:
mws.append(Descriptors.MolWt(mol))
print('number of compounds=%d' % len(mws))
print('min MW=%0.2f' % min(mws))
print('max MW=%0.2f' % max(mws))
print('average MW=%0.2f' % (sum(mws) / len(mws)))
number of compounds=858
min MW=129.17
max MW=2180.54
average MW=428.62
リピンスキーの法則によると、経口薬は分子量が500未満となる傾向があるとされています。これを信じるならば、このデータセットに含まれる化合物には、そのまま経口薬にするにはサイズが大きすぎるものも含まれていると言えそうです。
SMILESの読み込みとlogP予測
from rdkit import Chem
from rdkit.Chem import Crippen
smileses = ["CO", "CCO", "CCCO", "CCCCO"]
for smiles in smileses:
mol = Chem.MolFromSmiles(smiles)
logp = Crippen.MolLogP(mol)
print("%s:%0.2f" % (smiles, logp))
CO:-0.39
CCO:-0.00
CCCO:0.39
CCCCO:0.78
logP1そのものは、実験的に決定される数値で、簡単に言えば疎水性の指標になる値です。RDKitには、logPを計算によって予測するアルゴリズム2が実装されており、それがここで使用したMolLogP関数です。そして、実行結果からSMILESで表記されたアルキルアルコールのアルキル基部分が長くなるほど疎水性が高まるという予測結果になったことが分かります。ヒドロキシル基に比べてアルキル基が疎水的な性質を有していることを考えると、この結果は化学的に合理的な予測結果と言えるでしょう。
-
https://ja.wikipedia.org/wiki/%E5%88%86%E9%85%8D%E4%BF%82%E6%95%B0 ↩
-
S. A. Wildman and G. M. Crippen JCICS 39 868-873 (1999) ↩