はじめに
気軽に編集可能な(ベクター形式の)遺伝子位置の図を作りたくて、いろいろ探した結果、「これがいちばんかなあ、、(しぶしぶ)」という感じで使っているBiopythonの使い方です。
ほとんど同じことが、Biopythonのチュートリアルに書いてあリますが、自分の備忘録もかねてまとめました。
本家
http://biopython.org/DIST/docs/tutorial/Tutorial.html#htoc277
本家は、はgenbank形式のファイルからアノテーションをつけてますが、
gff3でも同じことができそうだったので、やったらできたので、その方法で書いておきます。
染色体用のですが、もちろんコンティグや特定領域の遺伝子構造も描画できます。
イントロン部分を線でつなぐ機能がないのが玉に瑕です。
公式チュートのデータを使うとこんな図ができます。
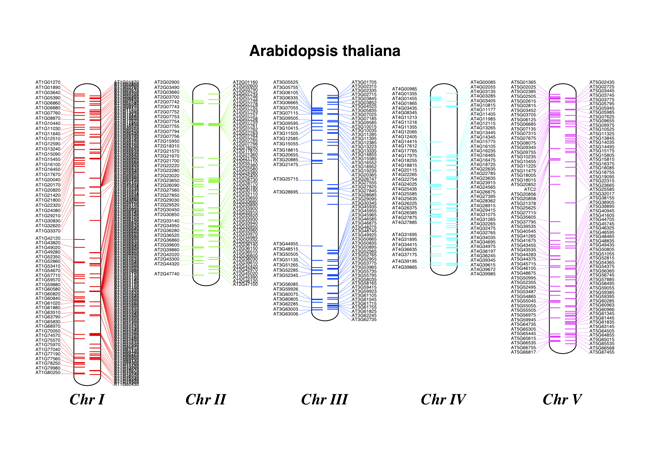
染色体だけ書くとき
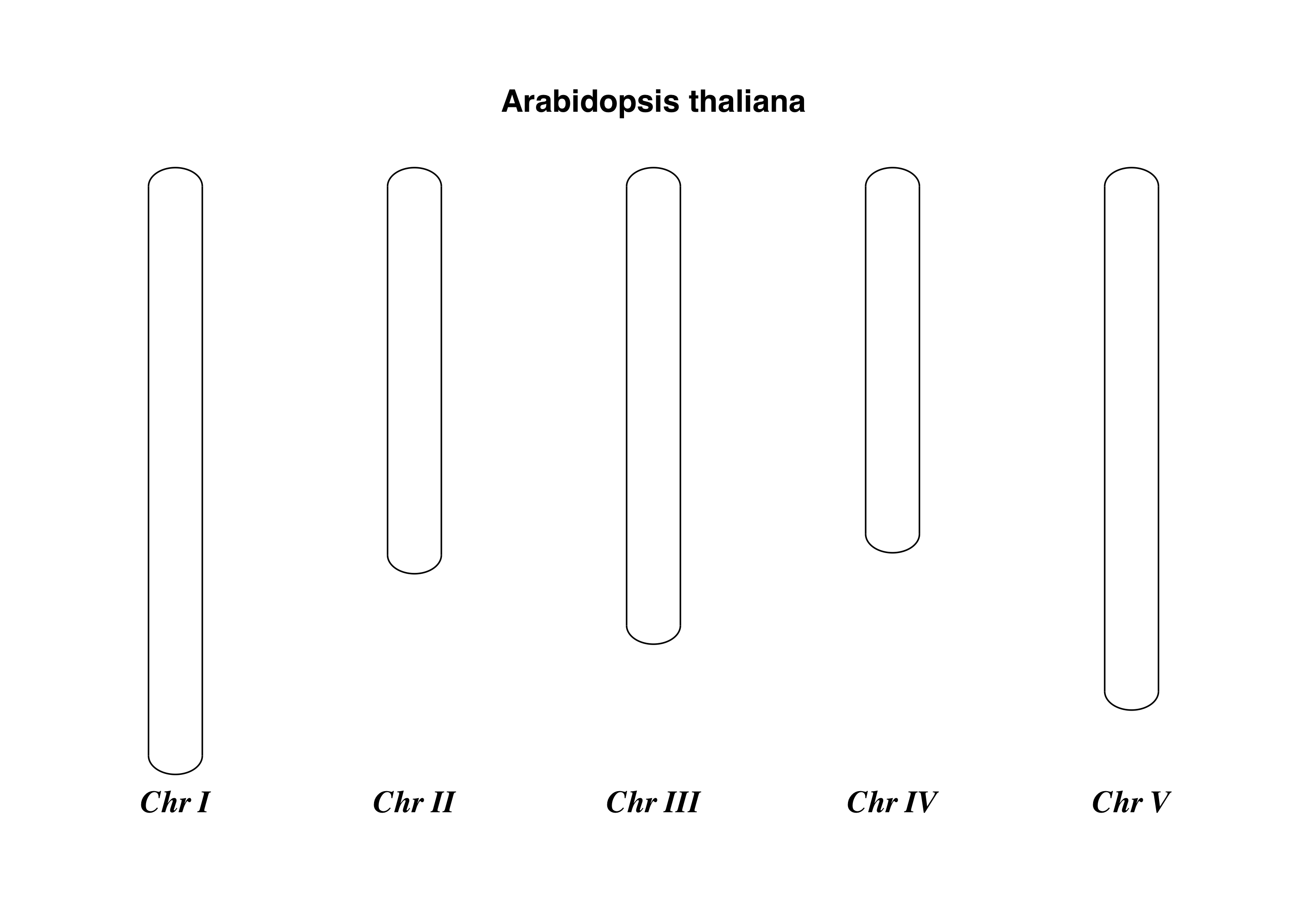
まず、アノテーションの無い、シンプルな染色体図を書いてみる
# !/usr/bin/env python
# -*- coding: utf-8 -*-
from reportlab.lib.units import cm
from Bio.Graphics import BasicChromosome
# 描画したい染色体の情報をリスト形式で入れる。染色体の名前、と配列全長(bp)
entries = [("Chr I", 30432563),
("Chr II", 19705359),
("Chr III", 23470805),
("Chr IV", 18585042),
("Chr V", 26992728)]
max_len = 30432563 #描画領域の領域を指定する。ここでは最大染色体長さに合わせてある。
telomere_length = 1000000 # 染色体の端の丸まってる部分はテロメアを示す(らしい)。その丸める領域を指定する。
chr_diagram = BasicChromosome.Organism() #描画オブジェクトを生成する(まだ空)
chr_diagram.page_size = (29.7*cm, 21*cm) # 描画エリアを指定するA4 landscape
for name, length in entries: #最初に作ったリストから情報を読み込んでいく
cur_chromosome = BasicChromosome.Chromosome(name) #nameの名前の染色体を1本作ってねと指定する
# Set the scale to the MAXIMUM length plus the two telomeres in bp,
# want the same scale used on all five chromosomes so they can be
# compared to each other
cur_chromosome.scale_num = max_len + 2 * telomere_length #染色体の最大の長さを指定する(丸める分を追加している)
# Add an opening telomere
start = BasicChromosome.TelomereSegment() #頭の方のテロメア(丸める)領域を指定している。
start.scale = telomere_length
cur_chromosome.add(start)
# Add a body - using bp as the scale length here.
body = BasicChromosome.ChromosomeSegment()#染色体本体部分の長さを指定している。
body.scale = length
cur_chromosome.add(body)
# Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True) #お尻の方のテロメア(丸める)領域を指定している。
end.scale = telomere_length
cur_chromosome.add(end)
# This chromosome is done
chr_diagram.add(cur_chromosome) #作った染色体オブジェクトを描画オブジェクトに追加している
chr_diagram.draw("simple_chrom.gif", "Arabidopsis thaliana") #pdf形式で出力させる。もちろん拡張子を変えれば形式は変えられる、、と書いたけど、pdfだけみたい。すいません。。最後に染色体群全体の名前を指定(今回はアラビ)している。
続けて、以上のスクリプトを、染色体長をfastaから直接取るように変更してみる。
# モジュールを読み込む
from reportlab.lib.units import cm
from Bio.Graphics import BasicChromosome
from Bio import SeqIO
# 描画したい染色体の情報をリスト形式で入れる。染色体の名前、該当するfastaファイルのパス を入れる
entries = [("Chr I", "Chr1.fa"),
("Chr II", "Chr2.fa")]
max_len = 30432563 #描画領域の領域を指定する。ここも最大長をfastaから求める様に変えてもよいが、今回は手打ちする
telomere_length = 1000000
chr_diagram = BasicChromosome.Organism() #描画オブジェクトを生成する(まだ空)
chr_diagram.page_size = (29.7*cm, 21*cm) # 描画エリアを指定するA4 landscape
# fastaから長さを求める部分を追加する。
for index, (name, fasta_filename) in enumerate(entries):
record = SeqIO.read(fasta_filename,"fasta") #seqIOを使って配列データを取り込む
length = len(record) #配列長を取得する
#ここ以降はほとんど一緒
cur_chromosome = BasicChromosome.Chromosome(name) #nameの名前の染色体を1本作ってねと指定する
# Set the scale to the MAXIMUM length plus the two telomeres in bp,
# want the same scale used on all five chromosomes so they can be
# compared to each other
cur_chromosome.scale_num = max_len + 2 * telomere_length
# Add an opening telomere
start = BasicChromosome.TelomereSegment() #頭の方のテロメア(丸める)領域を指定している。
start.scale = telomere_length
cur_chromosome.add(start)
# Add a body - using bp as the scale length here.
body = BasicChromosome.ChromosomeSegment() #染色体本体部分の長さを指定している。
body.scale = length #ここのlengthにfastaからとった長さが入るようになっている。
cur_chromosome.add(body)
# Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True) #お尻の方のテロメア(丸める)領域を指定している。
end.scale = telomere_length
cur_chromosome.add(end)
# This chromosome is done
chr_diagram.add(cur_chromosome) #作った染色体オブジェクトを描画オブジェクトに追加している
chr_diagram.draw("simple_chrom.pdf", "Arabidopsis thaliana")
遺伝子領域を描画する
染色体上に遺伝子の領域を描画する。
長さはfastaからとって、遺伝子情報はgff3からとるものとする。
# モジュールを読み込む
from reportlab.lib.units import cm
from Bio.Graphics import BasicChromosome
from Bio import SeqIO
from BCBio import GFF
# 描画したい染色体の情報をリスト形式で入れる。染色体の名前、該当するfastaファイルのパス、gffファイルのパス を入れる
entries = [("Chr I", "Chr1.fa", "Chr1.gff"),
("Chr II", "Chr2.fa", "Chr2.gff")]
max_len = 30432563
telomere_length = 1000000
chr_diagram = BasicChromosome.Organism()
chr_diagram.page_size = (29.7*cm, 21*cm)
# fastaから長さを求める部分を追加する。
for index, (name, fasta_filename, gff_filename) in enumerate(entries):
record = SeqIO.read(fasta_filename,"fasta")
length = len(record) #配列長を取得する
# gffからfeatureを取得する。
gff_handle = open(gff_filename)
for rec in GFF.parse(gff_handle):
features = [f for f in rec.features]
cur_chromosome = BasicChromosome.Chromosome(name)
cur_chromosome.scale_num = max_len + 2 * telomere_length
# Add an opening telomere
start = BasicChromosome.TelomereSegment()
start.scale = telomere_length
cur_chromosome.add(start)
# gff得たfeature情報を追加する
# Add a body - using bp as the scale length here.
body = BasicChromosome.AnnotatedChromosomeSegment(length, features) #<-ここ!
body.scale = length
cur_chromosome.add(body)
# Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True)
end.scale = telomere_length
cur_chromosome.add(end)
# This chromosome is done
chr_diagram.add(cur_chromosome)
chr_diagram.draw("simple_chrom.pdf", "Arabidopsis thaliana")
描画例
サンプルデータと描画例です
>contig1
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
contig1 test gene 200 201 3280.6 - . locus_tag=test1
contig1 test gene 400 800 3280.6 - . locus_tag=test2
contig1 test gene 800 1000 3280.6 - . locus_tag=test4
contig1 test gene 1500 1700 3267.1 - . locus_tag=test5
contig1 test gene 200 201 3280.6 - . locus_tag=test1
contig1 test gene 1500 1700 3267.1 - . locus_tag=test5
# !/usr/bin/env python
# -*- coding: utf-8 -*-
# モジュールを読み込む
from reportlab.lib.units import cm
from Bio.Graphics import BasicChromosome
from Bio import SeqIO
from BCBio import GFF
# 描画したい染色体の情報をリスト形式で入れる。染色体の名前、該当するfastaファイルのパス、gffファイルのパス を入れる
entries = [("Chr I", "test.fa", "test.gff"),
("Chr II", "test.fa", "test2.gff")
]
max_len = 2500
telomere_length = 0
chr_diagram = BasicChromosome.Organism()
chr_diagram.page_size = (29.7*cm, 21*cm)
# fastaから長さを求める部分を追加する。
for index, (name, fasta_filename, gff_filename) in enumerate(entries):
record = SeqIO.read(fasta_filename,"fasta")
length = len(record) #配列長を取得する
# gffからfeatureを取得する。
gff_handle = open(gff_filename)
for rec in GFF.parse(gff_handle):
features = [f for f in rec.features]
cur_chromosome = BasicChromosome.Chromosome(name)
cur_chromosome.scale_num = max_len + 2 * telomere_length
# Add an opening telomere
start = BasicChromosome.TelomereSegment()
start.scale = telomere_length
cur_chromosome.add(start)
# gff得たfeature情報を追加する
# Add a body - using bp as the scale length here.
body = BasicChromosome.AnnotatedChromosomeSegment(length, features) #<-ここ!
body.scale = length
cur_chromosome.add(body)
# Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True)
end.scale = telomere_length
cur_chromosome.add(end)
# This chromosome is done
chr_diagram.add(cur_chromosome)
chr_diagram.draw("simple_chrom.pdf", "TEST")
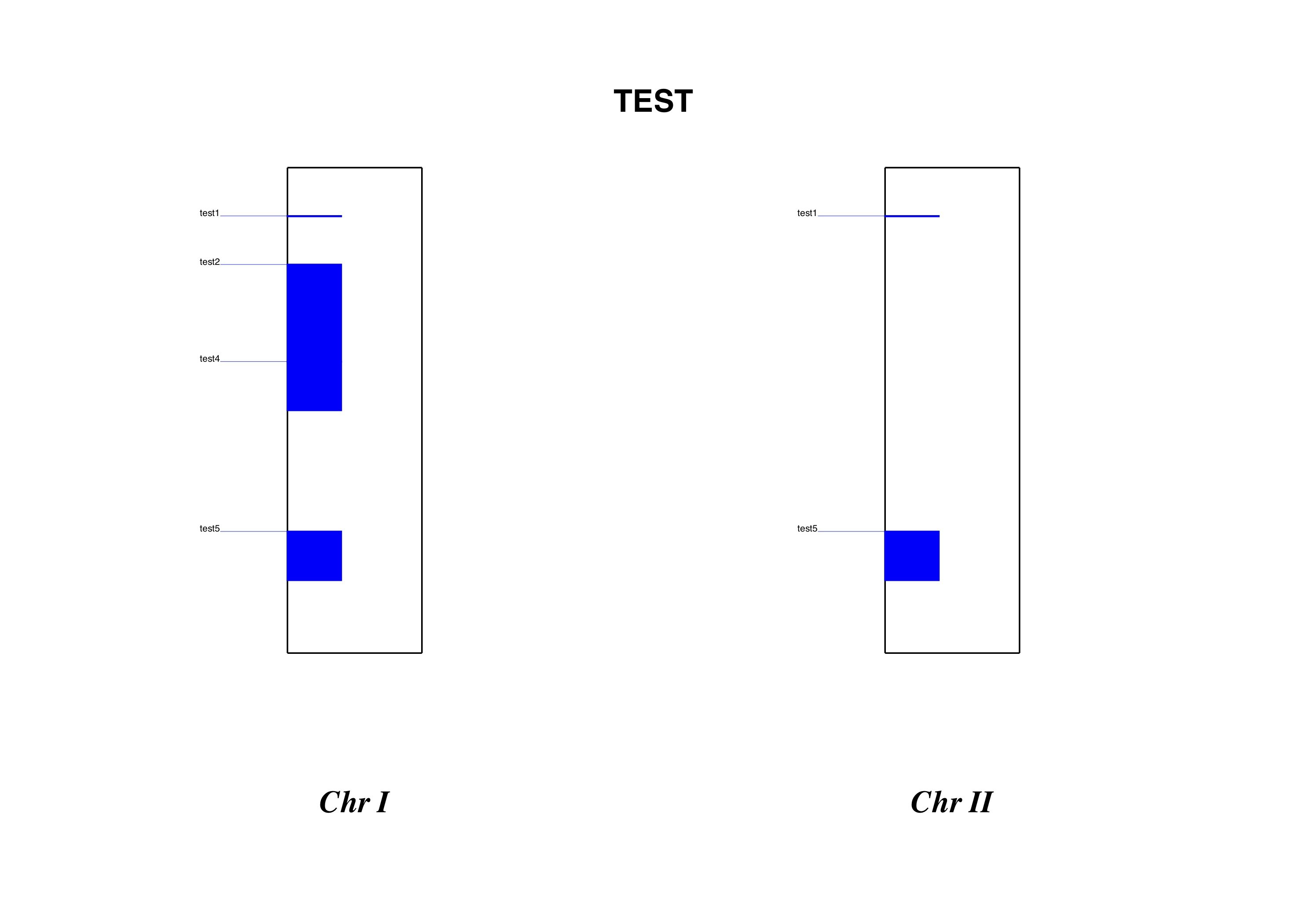
gffを使うときの注意
Parent項目がある場合、最も上位のParentに合わせて、すべての領域が遺伝子領域として描画される。
つまり3項目目がexonになっている行だけとってきたりしても、Parent=部分が残っているとexon-intronすべてが塗りつぶされる。これを避けるには、Parent=を除く。イントロン部分を細い線でつなぐ機能はない(と思う)。
各遺伝子のラベルにはlocus_tag=の内容が表示される。
スケールバーは表示されないので、自分が書きたい染色体 コンティグと別に、ラダーとなるような染色体を作ってスケールにするとよい。