これはR言語 Advent Calendar2025 の24日目の記事です。
はじめに
今回は、若干マニアックな内容になってしまうのですが、中学校で習ったメンデルの遺伝法則と高校で習うハーディーワインバーグの法則を使った簡単なシミュレーション実験をRでしてみます。
どんなことをシミュレーションしたいかというと、「近親交雑のもとではホモ接合をもつ個体の割合が増加し、結果的に分散が$(1+F_i)$倍される」ということです($F_i$は近交係数と呼ばれる値です。系譜を元に個体ごと決まります)。ホモ接合の個体が増えるのは感覚的にわかりますが、本当にそれが$(1+F_i)$倍されるのか、シミュレーションを使って検証していきたいと思います。
※ 私が博士課程で勉強している量的遺伝学にはゲノム関係行列と呼ばれる行列があり、その行列の対角成分は期待値として$1+F_i$になることが知られています。説明を読むとそれっぽいことは書いてあるのですが、いったい何を確率変数と見なしているのか、といった設定がいまいちよく分かりませんでした。たぶんこうだろうという設定が見つかったので、シミュレーションを通してそれを証明しようというのが今回の真の企みです。
シミュレーション設定
今回のシミュレーションでは、近親交雑が起こっていない場合と起こっている場合で比較を行いたいので、次の画像のような二つの系譜を用意しました。左のRandom Matingと書いてある方は近親交雑が起こっていない集団、右のInbreedingと書いてある方が起こっている集団です。人間で想像すると若干やばいですが、腹違いの子供同士($Y$と$Z$)が結婚して産んだ子供($X$)を考えている状況です。そして今回はこの$X$の分散を左と右で比較します。
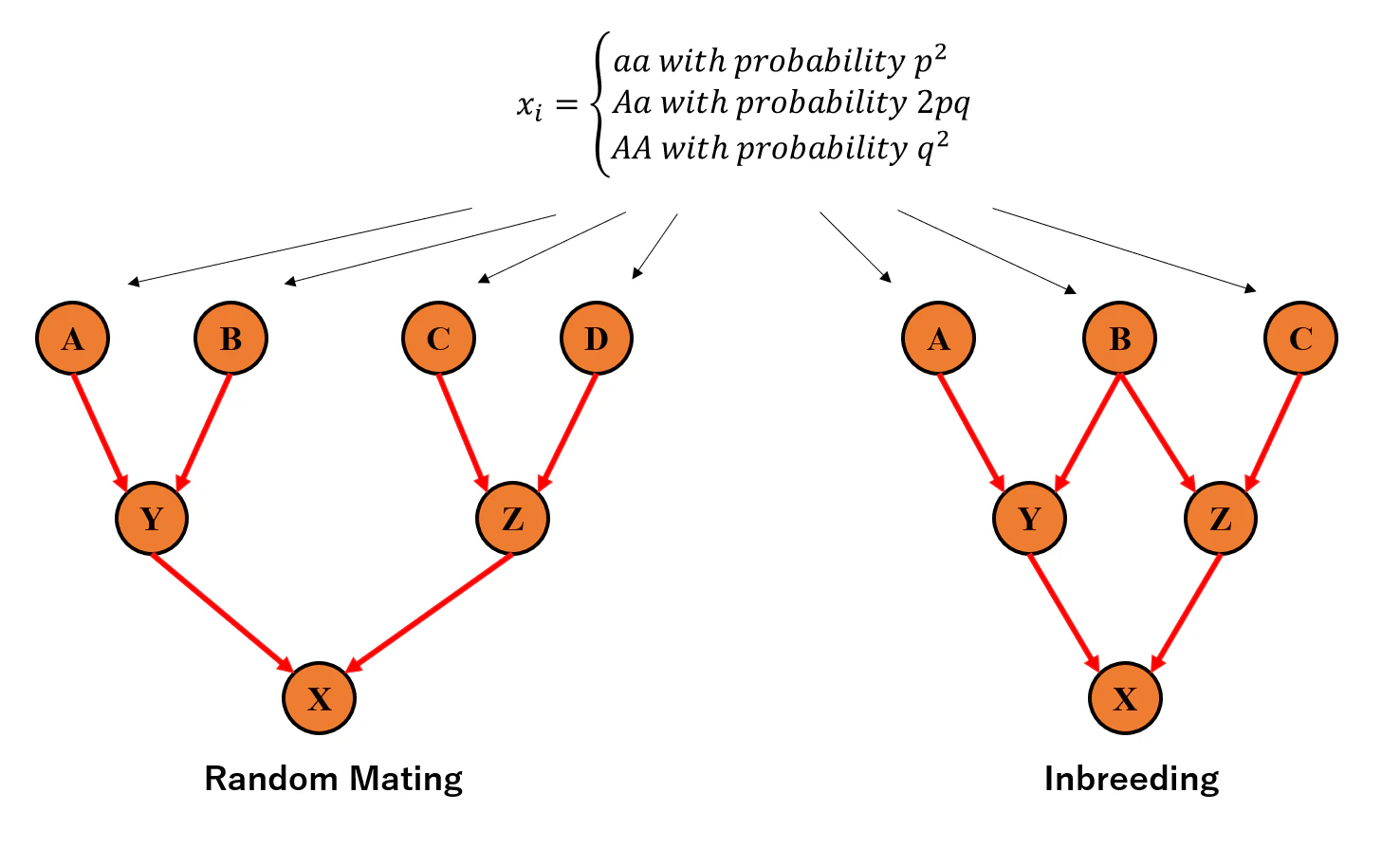
$X$が分散を持つ、というのが人によってはよくわからないと思いますが、ここがかなり重要なのでしっかり説明しようと思います。まずここで考えている$X$というのはある特定の遺伝子を持った個体ではなく、系譜のこの位置に存在しえるすべての個体を指しています。
例えば図の左の系譜において$A,B,C,D$の遺伝子型を$AA, Aa, aa, Aa$と仮にした時、それらは互いに交配しあって$X$が誕生します。この$X$の遺伝子型は確率的に$AA$だったり$Aa$だったり$aa$だったりしますので、それを元に分散を考えるということになります。また、今説明した分散は祖先集団$A,B,C,D$の遺伝子型を固定した上での条件付分散ですが、祖先集団の遺伝子型も確率的に決まってくるというのがさらに重要です(図の上を参照)。
従って、まずある確率に基づいて$A,B,C,D$の遺伝子型を決め、それに基づいて$Y,Z$さらに$X$を確率的に誕生させる、を何度も繰り返し、最後にその分散を取ることにすれば分散を計算できそうです(近交交雑のある図の右の系譜も同じ)。では実際にそれを実装していきます。
シミュレーション実装
よくやる設定ですがここからは$aa,Aa,AA$を、含んでいる$A$の数に基づいて$0,1,2$と符号化することにします。そして祖先集団の遺伝子型ですが集団における$a$の頻度を$p$、$A$の頻度を$q$とすると、$p+q=1$であり、$aa, Aa,AA$はそれぞれ$p^2,2pq,q^2$に従うことがハーディーワインバーグの法則から言えますので、今回はそのように祖先集団の遺伝子型を生成することとします。また今回は気分で$p=0.3$としました。
実際に書いたコードがこちらです。
p <- 0.3 #p=0.3で生成
q <- 1 - p #p+q=1よりq=0.7となる
# 交配用の関数。親の遺伝子型を入れるとメンデルの遺伝法則に基づいて確率的に子供の遺伝子型を返す
mate <- function(s, d) {
if (s == 0 & d == 0) {
return(0)
} else if (s == 2 & d == 2) {
return(2)
} else if ((s == 0 & d == 2) | (s == 2 & d == 0)) {
return(1)
} else if ((s == 1 & d == 0) | (s == 0 & d == 1)) {
return(sample(0:1, size = 1))
} else if ((s == 2 & d == 1) | (s == 1 & d == 2)) {
return(sample(1:2, size = 1))
} else if (s == 1 & d == 1) {
return(sample(0:2, size = 1, prob = c(0.25, 0.5, 0.25)))
}
}
# 左の系譜における$X$の遺伝子型を5000回発生
pedig1 <- sapply(1:5000, function(i) {
# 祖先集団の遺伝子型の生成。ベクトルの左からA,B,C,Dに対応
Origin <- sample(0:2, size = 4, prob = c(p^2, 2 * p * q, q^2), replace = TRUE)
# AとB,CとDを交配させた子供(XとY)をさらに交配し、その結果を返す
return(mate(mate(Origin[1], Origin[2]), mate(Origin[3], Origin[4])))
})
# 右の系譜における$X$の遺伝子型を5000回発生
pedig2 <- sapply(1:5000, function(i) {
# 祖先集団の遺伝子型の生成。ベクトルの左からA,B,Cに対応
Origin <- sample(0:2, size = 3, prob = c(p^2, 2 * p * q, q^2), replace = TRUE)
# AとB,BとCを交配させた子供(XとY)をさらに交配し、その結果を返す
return(mate(mate(Origin[1], Origin[2]), mate(Origin[2], Origin[3])))
})
# (i-1)番目の要素には1~i番目までのサンプルの分散が入る。
pedig1CumVar <- sapply(2:length(pedig1), function(i) {
return(var(pedig1[1:i]))
})
# (i-1)番目の要素には1~i番目までのサンプルの分散が入る。
pedig2CumVar <- sapply(2:length(pedig2), function(i) {
return(var(pedig2[1:i]))
})
# 結果のプロット まずは左の系譜の結果を黒で表示
plot(
pedig1CumVar,
pch = 19,
cex = 0.5,
col = 1,
ylim = c(0.35, 0.55),
ylab = "Estimated_Variance"
)
# 理論的に期待される分散
abline(h = 2 * p * q)
# 結果のプロット 右の系譜の結果を赤で表示
points(pedig2CumVar, pch = 19, cex = 0.5, col = 2)
# 理論的に期待される分散。近交係数は理論的に1/8となる。
abline(h = 2 * p * q * (1 + 1 / 8), col = 2)
# 凡例の追加
legend(
x = "topright",
legend = c("Random_Mate", "Inbreed"),
col = 1:2,
pch = 19
)
そしてこちらが出力されたプロットになります。
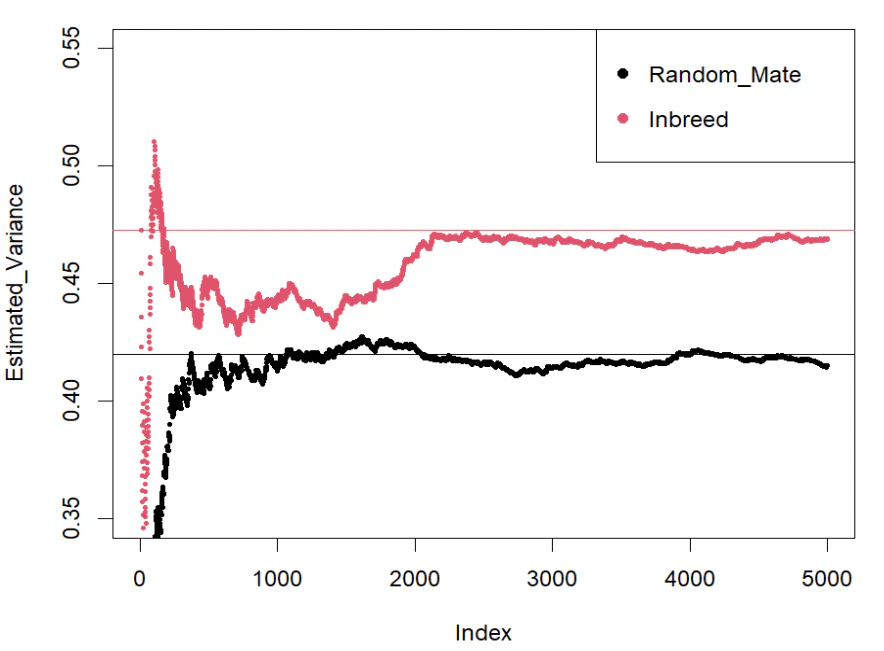
おもちゃみたいに簡単なシミュレーションなんですが、確かにそれぞれ理論値に収束していくことが分かりました。だいぶ収束が遅いの点は気になりますが…
まとめ
ということで今回は近交交雑のある集団とない集団でその分散がそれぞれ違ってくるのが、またその比は$(1+F_i)$になるのか、ということをシミュレーションを通して確認しました。ここら辺の話題は量定期遺伝学においてはなかなかイメージをするのが難しいものに分類されると思いますので、この記事がその理解の一助になればなによりです。