概要
PySBとはSystemBiologyを扱うためのPythonライブラリである。
document: https://pysb.readthedocs.io/en/stable/index.html
repository: https://github.com/pysb/pysb
今回、そのexamplesの一部の内容を確認したので、
自分のために簡易的にまとめておく。
(次回はPySBのexamplesを確認してみる(第2回 bax_pore_sequential.py & run_bax_pore_sequential.py)。)
環境
$ python --version
Python 3.10.0
$ pip freeze | grep pysb
pysb==1.13.2
(なお、インストールはpip install pysbで大まかになんとかなる)
内容
コード内容(bax_pore.py)
コード内容(bax_pore.py)
"""A version of BAX pore assembly where the subunits dimerize and then
tetramerize instead of assembling sequentially (contrast with
bax_pore_sequential.py). Inhibition of pore formation by Mcl-1 is also
implemented.
"""
from __future__ import print_function
from pysb import *
Model()
# Each BAX-BAX bond must always involve a t1 site on one monomer and a
# t2 site on the other.
Monomer('BAX', ['t1', 't2', 'inh'])
Annotation(BAX, 'http://identifiers.org/uniprot/Q07812')
Monomer('MCL1', ['b'])
Annotation(MCL1, 'http://identifiers.org/uniprot/Q07820')
# Two lone monomers form a dimer.
Parameter('kdimf', 1e-6)
Parameter('kdimr', 1e-7)
Rule('bax_dim',
BAX(t1=None, t2=None) + BAX(t1=None, t2=None) |
BAX(t1=1, t2=None) % BAX(t1=None, t2=1),
kdimf, kdimr)
# Two lone dimers form a tetramer, with a higher rate than the dimerization.
Parameter('ktetf', 1e-3)
Parameter('ktetr', 1e-4)
Rule('bax_tet',
BAX(t1=1, t2=None) % BAX(t1=None, t2=1) + BAX(t1=2, t2=None) % BAX(t1=None, t2=2) |
BAX(t1=1, t2=3) % BAX(t1=4, t2=1) % BAX(t1=2, t2=4) % BAX(t1=3, t2=2),
ktetf, ktetr)
# An inhibitory protein can bind to a BAX subunit at any time.
Parameter('kbaxmcl1f', 1e-5)
Parameter('kbaxmcl1r', 1e-6)
Rule('bax_inh_mcl1',
BAX(inh=None) + MCL1(b=None) |
BAX(inh=1) % MCL1(b=1),
kbaxmcl1f, kbaxmcl1r)
# Initial conditions
Parameter('BAX_0', 8e4)
Initial(BAX(t1=None, t2=None, inh=None), BAX_0)
Parameter('MCL1_0', 2e4)
Initial(MCL1(b=None), MCL1_0)
for p in BAX_0, MCL1_0:
Annotation(p, 'http://identifiers.org/doi/10.1371/journal.pcbi.1002482',
'isDescribedBy')
# We must fully specify all four BAX-BAX bonds, otherwise the pattern
# is too loose, match a given species multiple times (beyond the
# factor of four expected due to the rotational symmetry of the
# tetramer), resulting in erroneously high values.
Observable('BAX4', BAX(t1=1, t2=3) % BAX(t1=4, t2=1) % BAX(t1=2, t2=4) % BAX(t1=3, t2=2))
# Same all-bonds requirement here. However since the BAX tetramer is
# considered inhibited when even one subunit has an inhibitor bound,
# we only need to explicitly write inh=ANY on one of the monomer
# patterns.
Observable('BAX4_inh', BAX(inh=ANY, t1=1, t2=3) % BAX(t1=4, t2=1) % BAX(t1=2, t2=4) % BAX(t1=3, t2=2))
if __name__ == '__main__':
print(__doc__, "\n", model)
print("""
NOTE: This model code is designed to be imported and programatically
manipulated, not executed directly. The above output is merely a
diagnostic aid. Please see run_bax_pore.py for example usage.""")
コード内容(run_bax_pore.py)
コード内容(run_bax_pore.py)
"""Simulate the bax_pore model and plot the results."""
from __future__ import print_function
import matplotlib.pyplot as plt
from numpy import linspace
from pysb.simulator import ScipyOdeSimulator
from bax_pore import model
t = linspace(0, 100)
print("Simulating...")
x = ScipyOdeSimulator(model).run(tspan=t).all
plt.plot(t, x['BAX4'])
plt.plot(t, x['BAX4_inh'])
plt.legend(['BAX4', 'BAX4_inh'], loc='upper left')
plt.show()
コード構成
bax_pore.pyでモデルを定義し、
run_bax_pore.pyでそのモデルをimportし、シミュレート・出力する。
生物学的解説(BAX, MCL1, Bax poreとは)
出典:Wikipedia: BAX(タンパク質)、MCL1、Bax Pore関連
- BAX
- アポトーシスの調節因子(促進)。タンパク質。
- 普段は細胞質に存在するが、アポトーシスシグナル伝達に伴って、ミトコンドリア膜などに移動。
- ミトコンドリアの電位依存性アニオンチャネルと相互作用し、膜に孔を形成する。(つまり死ぬ)
- Bcl-2-associated X protein (Bcl-2結合Xタンパク質)
- 今回のモデルでは、単体、二量体、四量体を形成する。
- MCL1
- アポトーシスの抑制因子。タンパク質。
- Induced myeloid leukemia cell differentiation protein
- 今回のモデルでは、Baxに結合することで、穴の形成を阻害する。
- BAX pore
- poreは孔
- 参考画像はこちら(https://www.nature.com/articles/s41598-017-02825-7/figures/1)
コード解説
単量体の定義(BAX, MCL1)
from pysb import *
Model()
# Each BAX-BAX bond must always involve a t1 site on one monomer and a
# t2 site on the other.
Monomer('BAX', ['t1', 't2', 'inh'])
Annotation(BAX, 'http://identifiers.org/uniprot/Q07812')
Monomer('MCL1', ['b'])
Annotation(MCL1, 'http://identifiers.org/uniprot/Q07820')
from pysb import *, Model()はお約束。
MonomerのBAXは3つの部位('t1', 't2', 'inh')を持つ。
MonomerのMCL1は1つの部位('b')を持つ。
Annotation()でアノテーションを入れられる。
二量体形成(単量体+単量体=二量体)
# Two lone monomers form a dimer.
Parameter('kdimf', 1e-6)
Parameter('kdimr', 1e-7)
Rule('bax_dim',
BAX(t1=None, t2=None) + BAX(t1=None, t2=None) |
BAX(t1=1, t2=None) % BAX(t1=None, t2=1),
kdimf, kdimr)
片方の't1'と片方の't2'が結合する。
kdimf, kdimrのkは確かドイツ語由来だった気がする。dimはdimer(二量体)から、
f, rはおそらくforward, backwardから。
四量体形成(二量体+二量体=四量体)
# Two lone dimers form a tetramer, with a higher rate than the dimerization.
Parameter('ktetf', 1e-3)
Parameter('ktetr', 1e-4)
Rule('bax_tet',
BAX(t1=1, t2=None) % BAX(t1=None, t2=1) + BAX(t1=2, t2=None) % BAX(t1=None, t2=2) |
BAX(t1=1, t2=3) % BAX(t1=4, t2=1) % BAX(t1=2, t2=4) % BAX(t1=3, t2=2),
ktetf, ktetr)
空いていた't1','t2'も結合に繋がり塞がることになる。
四量体は英語ではtetramer。
なお、二量体形成反応より大分早い反応となっている。
抑制蛋白質の結合(BAX + MCL1 = BAX・MCL1)
# An inhibitory protein can bind to a BAX subunit at any time.
Parameter('kbaxmcl1f', 1e-5)
Parameter('kbaxmcl1r', 1e-6)
Rule('bax_inh_mcl1',
BAX(inh=None) + MCL1(b=None) |
BAX(inh=1) % MCL1(b=1),
kbaxmcl1f, kbaxmcl1r)
抑制蛋白MCL1は、BAXの'inh'にいつでも結合できる。
初期条件の指定
# Initial conditions
Parameter('BAX_0', 8e4)
Initial(BAX(t1=None, t2=None, inh=None), BAX_0)
Parameter('MCL1_0', 2e4)
Initial(MCL1(b=None), MCL1_0)
for p in BAX_0, MCL1_0:
Annotation(p, 'http://identifiers.org/doi/10.1371/journal.pcbi.1002482',
'isDescribedBy')
初期条件BAX_0とMCL1_0を定義し登録している。Annotationもつけている。
観察対象の指定
Observable('BAX4', BAX(t1=1, t2=3) % BAX(t1=4, t2=1) % BAX(t1=2, t2=4) % BAX(t1=3, t2=2))
Observable('BAX4_inh', BAX(inh=ANY, t1=1, t2=3) % BAX(t1=4, t2=1) % BAX(t1=2, t2=4) % BAX(t1=3, t2=2))
観察対象を指定する。
-
BAX4:四量体 -
BAX4_inh:4つのうち一つでも'inh'にMCL1が結合している四量体
inh=ANYは、相手が誰でも結合はしている状態を示す。(cf. pysb.core.ANY)
モデルの読み込み
import matplotlib.pyplot as plt
from numpy import linspace
from pysb.simulator import ScipyOdeSimulator
from bax_pore import model
from bax_pore import modelで、
bax_pore.pyでの定義が自動的に格納されたmodelをimportできる。
シミュレート
t = linspace(0, 100)
print("Simulating...")
x = ScipyOdeSimulator(model).run(tspan=t).all
ScipyOdeSimulator(model).run(tspan=t)でシミュレート。
結果の描画
plt.plot(t, x['BAX4'])
plt.plot(t, x['BAX4_inh'])
plt.legend(['BAX4', 'BAX4_inh'], loc='upper left')
plt.show()
出力
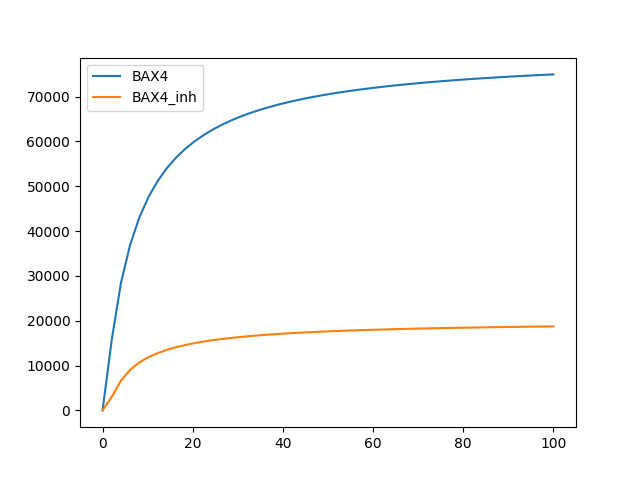
四量体形成が速いので四量体が蓄積される。
阻害部位への結合は、四量体形成を阻害するものではなく、BAXの状態に関係なく作用するので、大体一定の割合を占める。(ネガティブフィードバックはないという程度の意味)
感想
書き写しながらまとめることで、目があまり滑らなくてよかった。
訳語や解説等は調整が必要かとは思うが、気が向いたら書き直そうと思う。
Annotation(), pysb.core.ANYが今回初めて知ったものだった。
出典等
文中参照