この記事はバイオインフォマティクス Advent Calendar 2021(13日目)に登録されています。
どういう記事か
AlphaFold2によるタンパク質立体構造予測や、Enformerによる遺伝子発現予測など、ここ数年でバイオインフォマティクス分野が急速に活発化しているのを感じています。「生物×情報科学って面白そう!」という方も、増えてきているのではないでしょうか。
この記事では下記の方々を対象として、最近の生物配列解析について軽く解説したいと思います。
1. 生物に馴染みの無い情報工学分野出身の方
2. 機械学習/深層学習に馴染みの無いウェット系ラボ出身の方
3. その他のバイオインフォマティクスに興味がある全ての方
1. Sequence
生体の最も基本的な構成要素は、脂質・タンパク質・糖・核酸(DNA/RNA)等の生体分子です。膨大な種類の生体分子の、様々な機能によって、あらゆる生命活動が行われています。
遺伝子は核酸(≒塩基)が繋がってできたものなので、塩基配列という文字列で表記できます。遺伝子を「翻訳」することでできるタンパク質も、アミノ酸が繋がったペプチド鎖が折り畳まってできたものなので、アミノ酸配列という文字列で表記できます。
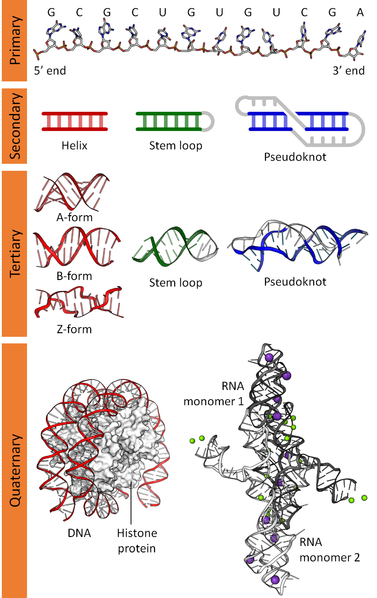
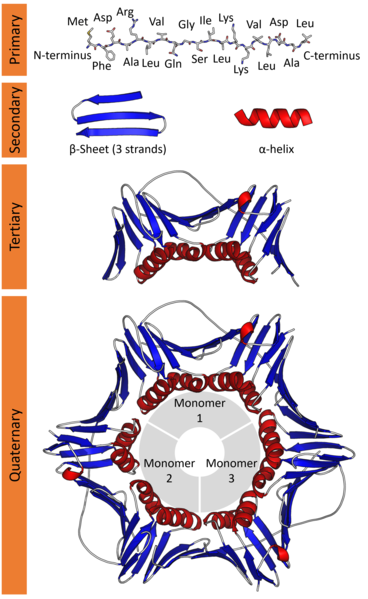
タンパク質はペプチド鎖が植物のツタのようにクルクル丸まってできた巨大分子です。その性質として、特定の低分子と好んで結合する**「鍵と鍵穴の関係」や、レゴブロックの組み立てのような「高次構造形成」**が知られています。これらは、タンパク質の複雑な立体構造によるものです。このような現象により、まるでピタゴラスイッチのように、特定のタイミングで特定の機能を有するタンパク質が活性化/不活性化され、生命活動の信号となります。
このような事情があって、タンパク質の機能、機能を決定付ける立体構造、立体構造を形作るアミノ酸配列、アミノ酸配列を指定するDNA配列、等をつぶさに調べれば、生命現象を理解することができるのではないか? という考えが生まれたのだと思います。
2. Alignment
配列解析の基本的なアルゴリズムのことで、アラインメント(あるいは単にアライメント)と読みます。複数の配列を一行の文字列として並べたときに、似ている配列パターンを重ね合わせるように整列させる操作です。
アミノ酸配列のアラインメント

突然変異によりDNA/アミノ酸の欠失や挿入が起こると、配列に違いが生じます。逆に言えば、異なる配列を比較したときに、同一の進化的起源を持つ配列パターンを特定することができます。このような配列パターン(モチーフ)は「進化的に保存」されており、生存や子孫繁栄に有利な機能を持つことが期待できます。また、突然変異の蓄積率を算出することで、元の配列がどのように進化の枝分かれを辿って行ったのかを調べることができます(分子系統解析)。
あるいは、構造的特徴そのものが機能に重要であることもあります。そうした場合は、配列が多く置換されていても立体構造が類似することがあります。実際に、一般に配列の保存性よりも構造の保存性が高いことがよく知られています。
3. Sequence Embedding
生物配列解析の王道は分子系統解析なのですが、本稿では、機械学習/深層学習を用いた生物配列解析について深堀していきたいと思います。
生物配列を機械に読み込ませるためには、文字列を何らかのベクトル表現に変換する必要があります (embedding)。AlphaFold2はアミノ酸配列を入力として立体構造を予測する深層学習モデルでした。ベクトル化のために配列のone-hot encodingを行い、文字列をバイナリ行列に変換しています1。
3.1 Protein Language Model(タンパク質言語モデル)
Arguably, proteins are the most important machinery of life. Protein sequence largely determines protein structure, which somehow determines protein function. Thus, the expression of the language of life are essentially protein sequences.2
最も有名なタンパク質言語モデルの一つが、Tasks Assessing Protein Embeddings(TAPE)です4。TAPEでは、5つの深層学習モデルを用いて5つのタンパク質物性予測タスクのベンチマークを行っています5。自然言語深層学習の急速な発展に伴って研究が進んでいる分野で、今年10月にはProteinBERTというTAPEを拡張したモデルも発表されました6。ここら辺の動向は木原先生(@d_kihara)らの総説がよくまとまっているので、詳細に知りたい方はご参考ください。
pip install bio-embeddings[all]
使用可能なモデルは以下の12個です。
- Fastext
- Glove
- Word2Vec
- SeqVec
- ProtTrans (ProtBert, ProtAlbert, ProtT5)
- UniRep
- ESM/ESM1b
- [PLUS] (https://github.com/mswzeus/PLUS/)
- [CPCProt] (https://www.biorxiv.org/content/10.1101/2020.09.04.283929v1.full.pdf)
- [PB-Tucker] (https://www.biorxiv.org/content/10.1101/2021.01.21.427551v1)
- [GoPredSim] (https://www.nature.com/articles/s41598-020-80786-0)
- [DeepBlast] (https://www.biorxiv.org/content/10.1101/2020.11.03.365932v1)
使い方はこんな感じで、とても使いやすそうです。継続的なアップデートに期待。
from bio_embeddings.embed import SeqVecEmbedder
embedder = SeqVecEmbedder()
embedding = embedder.embed("SEQVENCE")
print(embedding.shape)
#(3, 8, 1024)
3.2 Sequence Descriptor (配列記述子)
上記タンパク質言語モデルの他に、組成や物性から特徴量(sequence descriptor)を計算する古くからある方法も活用されています。アミノ酸記述子9としては、AAIndex10が有名です。
AAindexはアミノ酸およびアミノ酸の組を物理化学的・生化学的な記述子に変換するためのデータベースで、日本のバイオインフォマティクス研究の大御所、金久實先生の研究成果です。データは全て公開文献に由来していて、現在までに566個のアミノ酸記述子、94個の置換マトリクス(BLOSUM62等)、47個のタンパク質接触ポテンシャルが公開されています。
ケモインフォマティクス分野では、化合物の物理化学的性質を計算することができるRDKitというソフトウェアを用いることで、化合物データセットをいわゆるテーブルデータと呼ばれる構造化データに変換することができます。これにより、タイタニックの生存者予測と同じような機械学習を行うことができます。同様にアミノ酸記述子を用いることで、配列/アミノ酸から構造化データを生成してタンパク質の物性予測モデルを作ることができます。
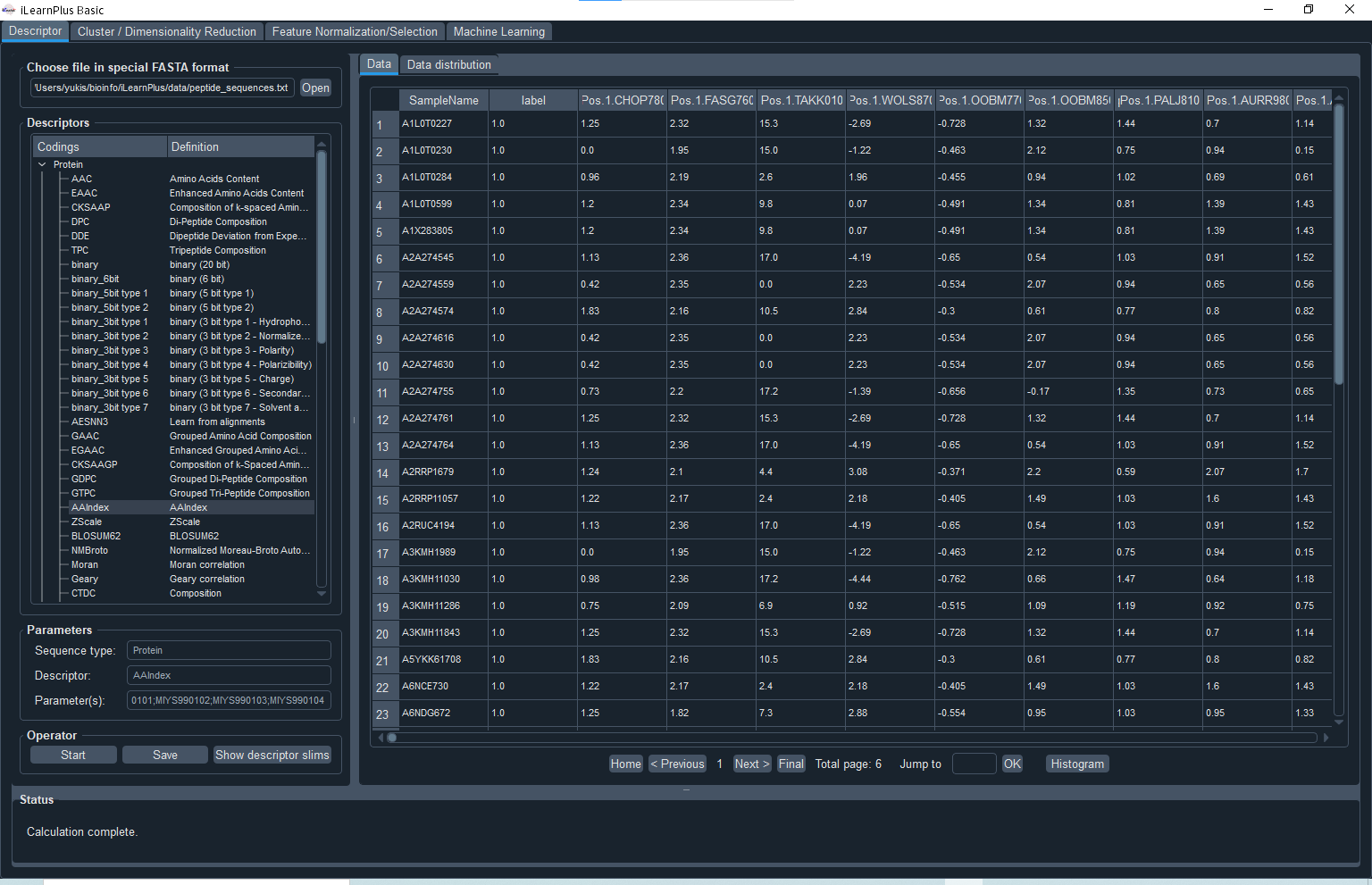
配列記述子を計算するソフトウェアは急激に増えてきている段階で、まだRDKitやMordred11のような信頼性のある整備されたものはほとんどないように思います。ただ、その中でも使いやすそうだなと個人的に感じたのはiLearnPlus12です。
iLearnPlusはGUIベースで配列記述子を生成できるpythonライブラリです。配列ファイル(FASTA形式)を開いた後は、記述子生成から機械学習モデル作成まで一気通貫で実行することができます。インストール方法も簡単で、python3.6以降のいくつか依存パッケージが入った適当なconda環境を用意して、下記コマンドを実行するだけです。
iLearnPlus$ python iLearnPlus.py
操作画面はこんな感じ。う~~ん、便利すぎる。
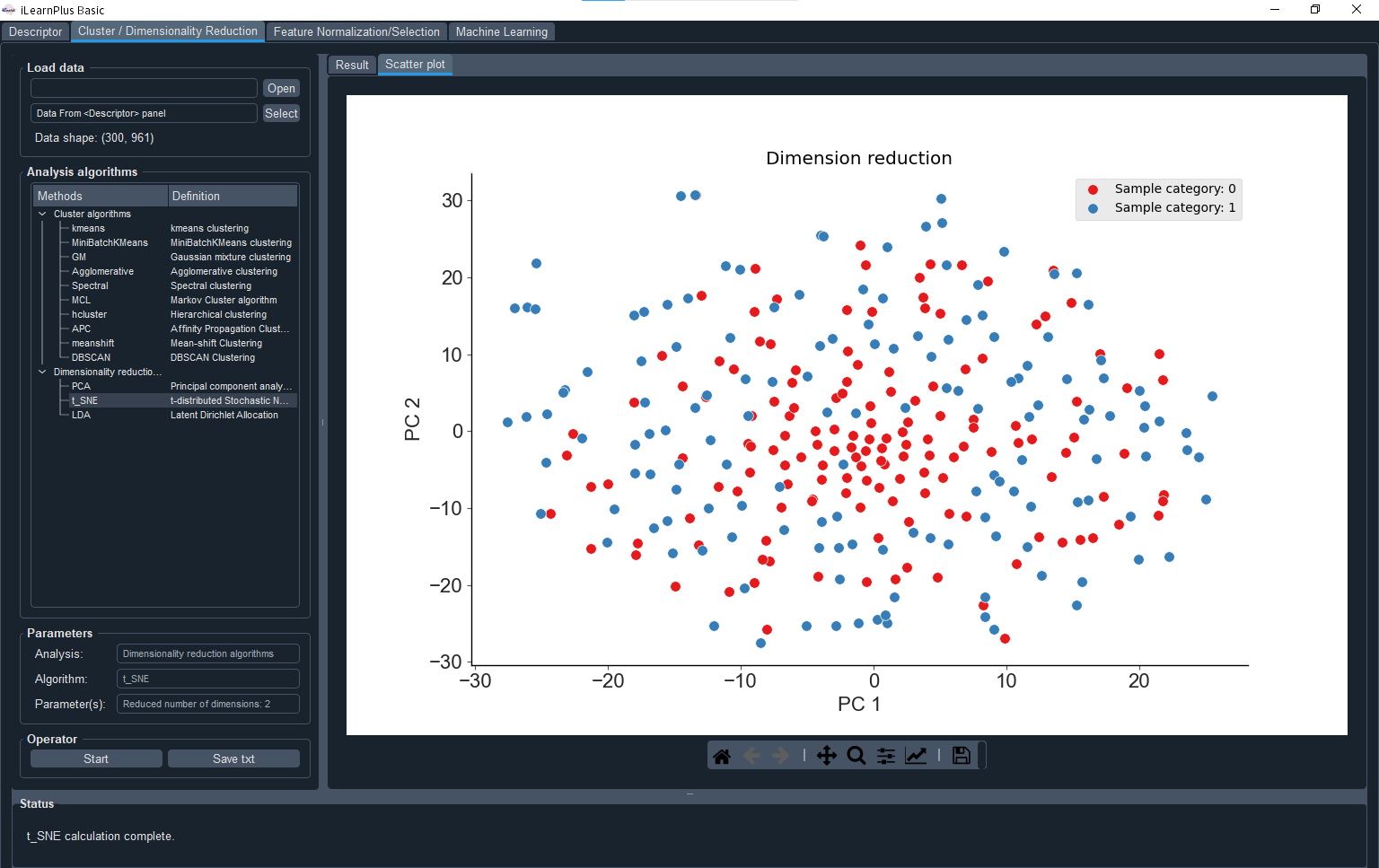
もちろん、生物配列であるDNA、RNA、アミノ酸どれでも使用可能です。
今年11月には、さらに発展的な記述子として数学的な変換を施した配列記述子MathFeature13も提案されています。
元論文 Figure1より引用

まとめ
機械学習/深層学習を適用する形で、生物配列解析も日進月歩で進化しています。巨大データベースの強みを生かした深層学習モデルだけが脚光を浴びるのではなく、古典的な配列記述子による解析も再評価・再注目されつつあるように感じます。参考文献のほとんどは2、3年以内に発表された論文で、改めて驚きました。そして思った以上に、簡単に色々な解析ができそうだと感じました。機会があればこれらのライブラリの詳細な使い方などを改めて紹介してみたいです。
最後までお読みいただき、ありがとうございました。これを読んだあなたはもうバイオインフォマティシャン! 一緒にこの分野を盛り上げていきませんか?
-
実際的には、進化的に保存された構造を抽出しやすくするために、アミノ酸残基の変異出現頻度を示すposition-specific score matrix (PSSM) という行列表現を用いた学習を行っています。
ただ、単なるバイナリ行列からは生物学的意味が掬い取りづらいため、PSSM、Word2Vecにおける単語埋め込みのようなベクトル表現、ケモインフォマティクス分野におけるグラフ表現や記述子生成、等の手法を用いるのが一般的なようです。 ↩ -
Heinzinger, M., Elnaggar, A., Wang, Y. et al. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinformatics, 20, 723 (2019). https://doi.org/10.1186/s12859-019-3220-8
配列は文字列であること、高次構造を形成することを述べました。極端な見方をすると、DNAやタンパク質は「文・単語・イディオム・文章」という階層構造を持った自然言語と類似した性質を持つとも言えます。このような考えから生まれたのがタンパク質言語モデルです3。 ↩ -
Hie B, Zhong ED, Berger B, Bryson B. Learning the language of viral evolution and escape. Science, 15, 371(6526):284-288 (2021). doi: 10.1126/science.abd7331. 『人間の言語から構築したAIモデルが、「ウイルスの変異」も予測する』 by WIRED. ↩
-
Rao R, Bhattacharya N, Thomas N, et al. Evaluating Protein Transfer Learning with TAPE. Adv Neural Inf Process Syst, 32, 9689-9701 (2019). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7774645/ ↩
-
モデルは、LSTM、bidirectional LSTM、unidirectional mLSTM、ResNet、Transformer。構造関連の予測として3つ(二次構造予測、アミノ酸残基間相互作用予測、遠縁関係にあるタンパク質の予測)。機能関連の予測として2つ(点変異による緑色蛍光タンパク質GFPの蛍光強度変化の予測、点変異による熱安定性変化の予測)。 ↩
-
新規タスクが5つ加えられ、モデルが軽量化されました。また、GOという配列に対する生物学的アノテーションで事前学習を行っている特徴があります。
こうした深層学習モデルの特徴は、膨大なデータでタンパク質物性を事前学習していることです。概して生化学実験が律速になりがちのバイオインフォマティクス分野において、少数サンプルで高い精度のタンパク質機能予測が可能であるという報告がされています7。 ↩ -
Biswas, S., Khimulya, G., Alley, E.C. et al. Low-N protein engineering with data-efficient deep learning. Nat Methods, 18, 389–396 (2021). https://doi.org/10.1038/s41592-021-01100-y
タンパク質言語モデルライブラリに簡易にアクセスできるライブラリとしてBio Embeddings8があり、pipで簡単にインストールできます。 ↩ -
Dallago, C., Schütze, K., Heinzinger, M., Olenyi, T., Littmann, M., Lu, A. X., Yang, K. K., Min, S., Yoon, S., Morton, J. T., & Rost, B. Learned embeddings from deep learning to visualize and predict protein sets. Current Protocols, 1, e113 (2021). doi: 10.1002/cpz1.113 ↩
-
造語です。Google翻訳で4件しかヒットしなかったのですが、amino acid descriptorsだと5000件くらいヒットしたので、未翻訳なだけかも。 ↩
-
Nakai, K., Kidera, A., and Kanehisa, M. Cluster analysis of amino acid indices for prediction of protein structure and function. Protein Eng, 2, 93-100 (1988). ↩
-
Moriwaki, H., Tian, YS., Kawashita, N. et al. Mordred: a molecular descriptor calculator. J Cheminform 10, 4 (2018). https://doi.org/10.1186/s13321-018-0258-y ↩
-
Zhen Chen, Pei Zhao, Chen Li, Fuyi Li, Dongxu Xiang, Yong-Zi Chen, Tatsuya Akutsu, Roger J Daly, Geoffrey I Webb, Quanzhi Zhao*, Lukasz Kurgan*, Jiangning Song*, iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Research, gkab122, (2021). https://doi.org/10.1093/nar/gkab122 ↩
-
Robson P Bonidia, Douglas S Domingues, Danilo S Sanches, André C P L F de Carvalho, MathFeature: feature extraction package for DNA, RNA and protein sequences based on mathematical descriptors. Briefings in Bioinformatics, bbab434, (2021). https://doi.org/10.1093/bib/bbab434 ↩