MZmine3はアメリカのグループが提供する質量分析データ解析用のソフトウェアである。メタボローム解析などに用いることができる。
基本的な使い方を以下のページの資料を元にまとめた。
以下の資料はMZmine2のものであるが、MZmine3にも相同のソフトが存在するため、web pageを参照に相同ソフトを選択し、利用するよう調整した。
本記事での解析は本格的なメタボローム解析は想定しておらず、2つの解析条件の比較から効率的に特徴的な差を見出すためのツールとしてMZmine3を用いている。また、対象となる化合物は未知化合物であることを仮定している
データの変換
mzML形式に変換するとよい。 様々な方法があるが、現状converterを使う必要がある。 いくつかのホームページでconverterに関して解説されている。MZmine3のページにおける解説
理研が開発したメタボロームツールMS-DIALのページにおける解説
MSconvertが比較的使いやすそうである。
このソフトウェアはWindowsにインストール可能であるため、インストールして利用する。
- プログラムを起動後、Browsボタンから目的のデータを選択する。
- Output formatでmzMLを選択する。
- Binary econding precisionは64 bitを選択する。
- Use zlib compressionにはcheckを入れた方がデータサイズを圧縮できる (しなくてもよい)
- peakPicking vendor msLevel=1-2 のフィルターを追加する。
- Startをクリックする。
データのインポート
mzML形式のファイルをウィンドウにドラッグアンドドロップをすることでデータを開くことができる。基本的なデータの観察
データを開くと以下のような画面になる。 左側のウィンドウにデータ名が表示される。
データ名をダブルクリックするとRaw dataタブが開き、クロマトグラムが右側のウィンドウに表示される。
下のウィンドウにはscan tableが表示される。
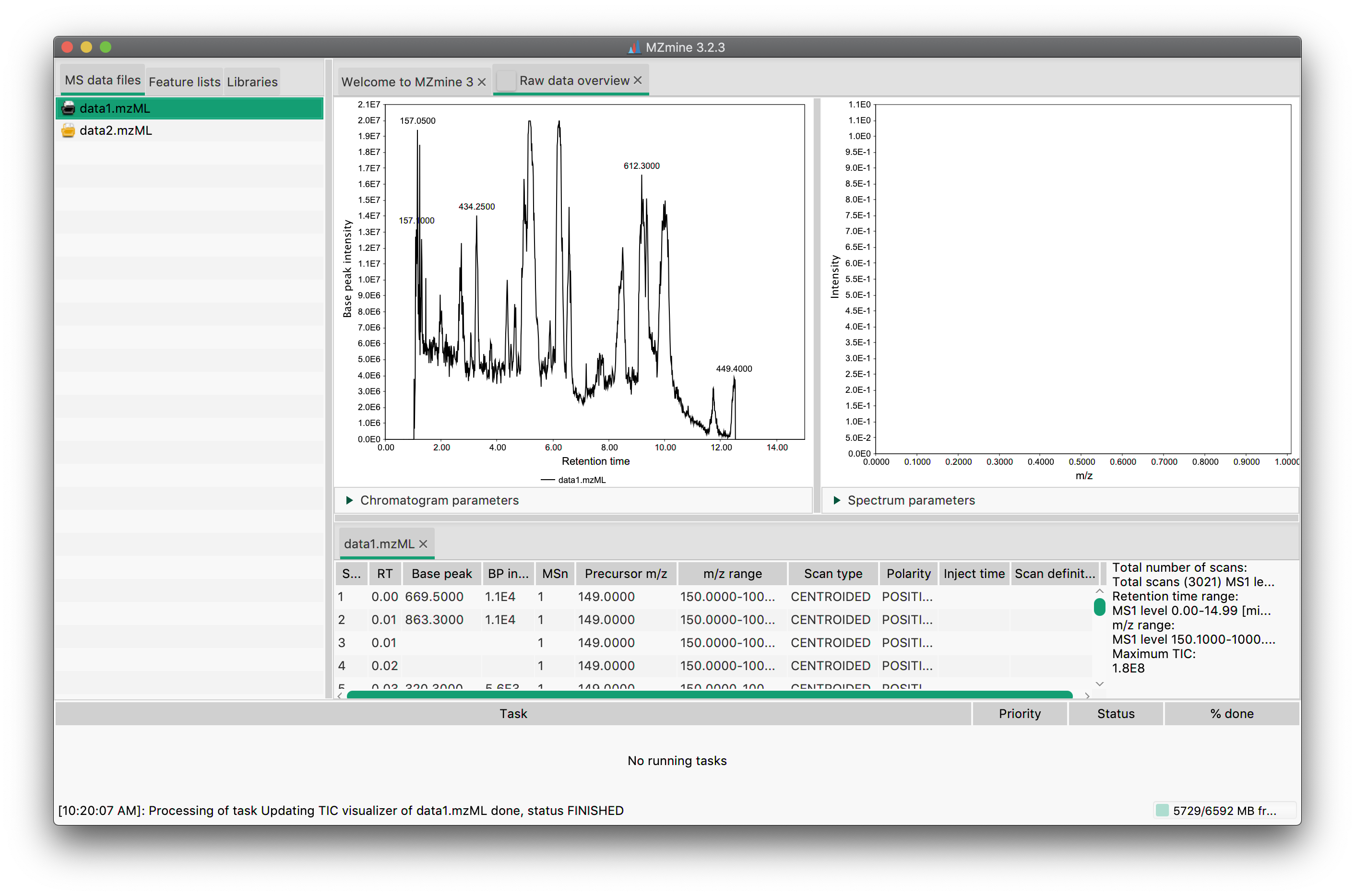
Raw data overviewのとなりのチェックボックスにチェックを入れ、複数データファイルを選択することで、複数のデータを同じウィンドウに表示させることができる。
クリックした箇所のスペクトルが右側のウィンドウに表示される。
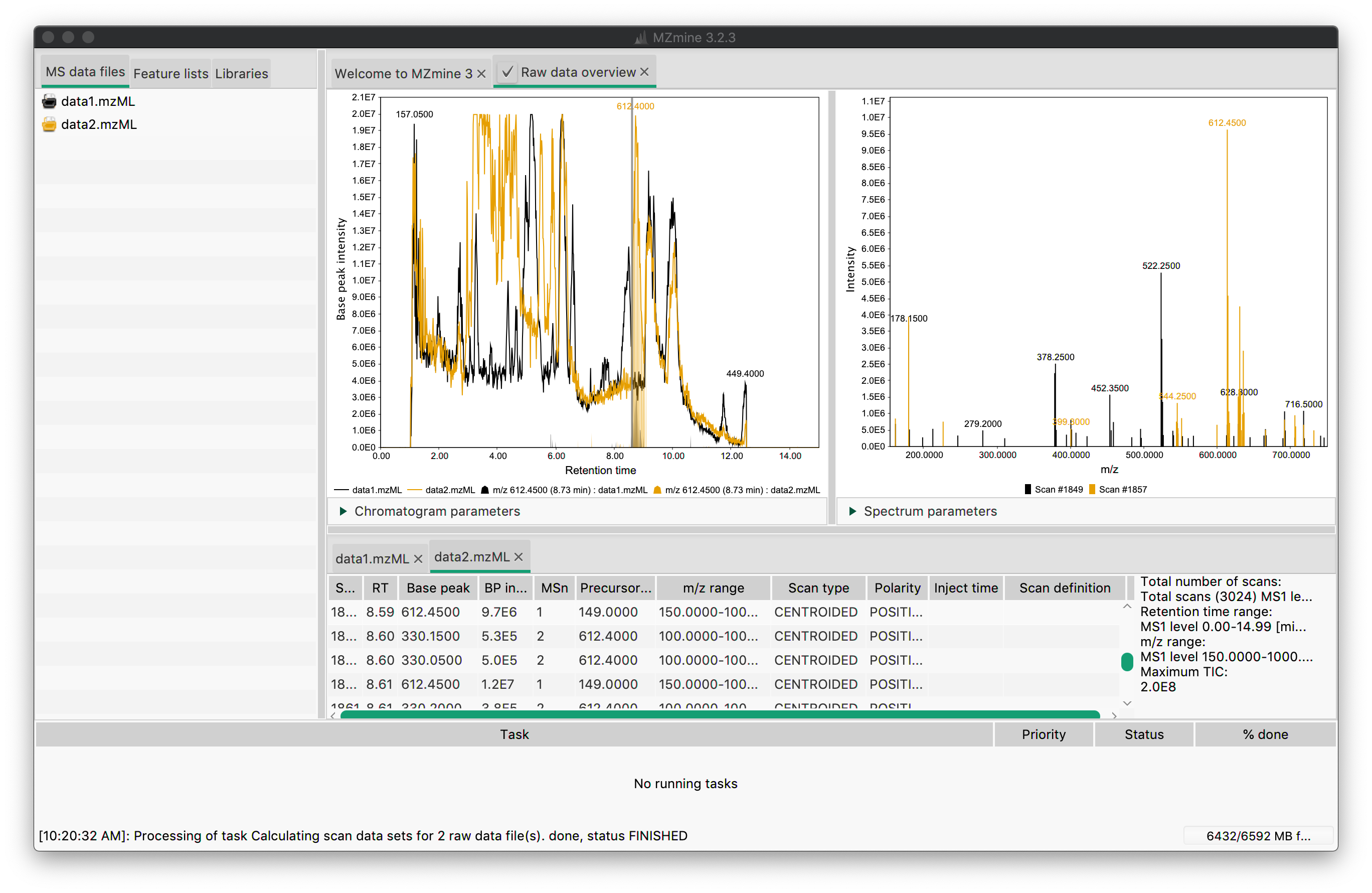
軸を左クリックし動かすことで表示範囲を動かすことができる。
また、軸の上でスクロールホイールを回すことで表示範囲の拡大縮小ができる。
また、ウィンドウ内をドラッグすることで、ドラッグした範囲を拡大することができる。
データ処理
2つの代謝データを比較する場合、以下の処理の結果出力されたListを比較するとよい。- Peak detection
- Chromatogram building
- Deconvolution
- 13C isotope filter
- Alignment
Peak detection
Raw data Methods > Mass detection > Mass detection を実行する。
データはcentroid形式である必要がある。
Mass detectorの行のSet upをクリックし、ノイズの閾値を設定する。
今回は「1.0E2」とした。
OKをクリックする。

Chromatogram building
Feature detection > LC-MS > ADAP Chromatogram builder を選択
Scan to scan accuracyは機械の性能に応じて設定する。

Feature Listsタブに結果が出力される。

Deconvolution
Feature detection > Chromatogram resolving > Local minimum resolver を実行
特に設定をいじらずに実行する。
Features listsに新しいデータが出力される。
データの重ね合わせによる確認
Features listsに新しく出てきたファイルをクリックする。
右側に出てきたリストを全て選択し、右クリックからshow XIC dialogをクリックする。

その後、よくわからない画面が出てくるが、ウィンドウ右端の鍵のマークをクリックすると以下のようなpeakとTICを重ね合わせた図を見ることができる。

13C isotope filter
まず、deconvolutionで出力されたFeature listsを選択する。
Feature list methods → Isotopes → 13C isotope filter を実行する。
Feature listsはas selected in main windowを選ぶ。
パラメーターの設定は目的のLCMSの性能に応じて設定する。

新しいFeature listsが出力される。
Identification
HR-MS測定データであれば化合物の同定をある程度行うことができる。Features listsから特定の化合物を選択し、右クリックからpredict molecular formulaを選ぶことで実行可能である。

Alignment
2つのデータ間を比較し、同じ化合物をグルーピングすることができる。Feature list methods → Alignment → Join aligner を実行する。
必要に応じてパラメーターを変更する。
今回し用したMSは分解能が高くないので、m/z toleranceを緩めに設定しています (デフォルトでは20 ppmです。)。
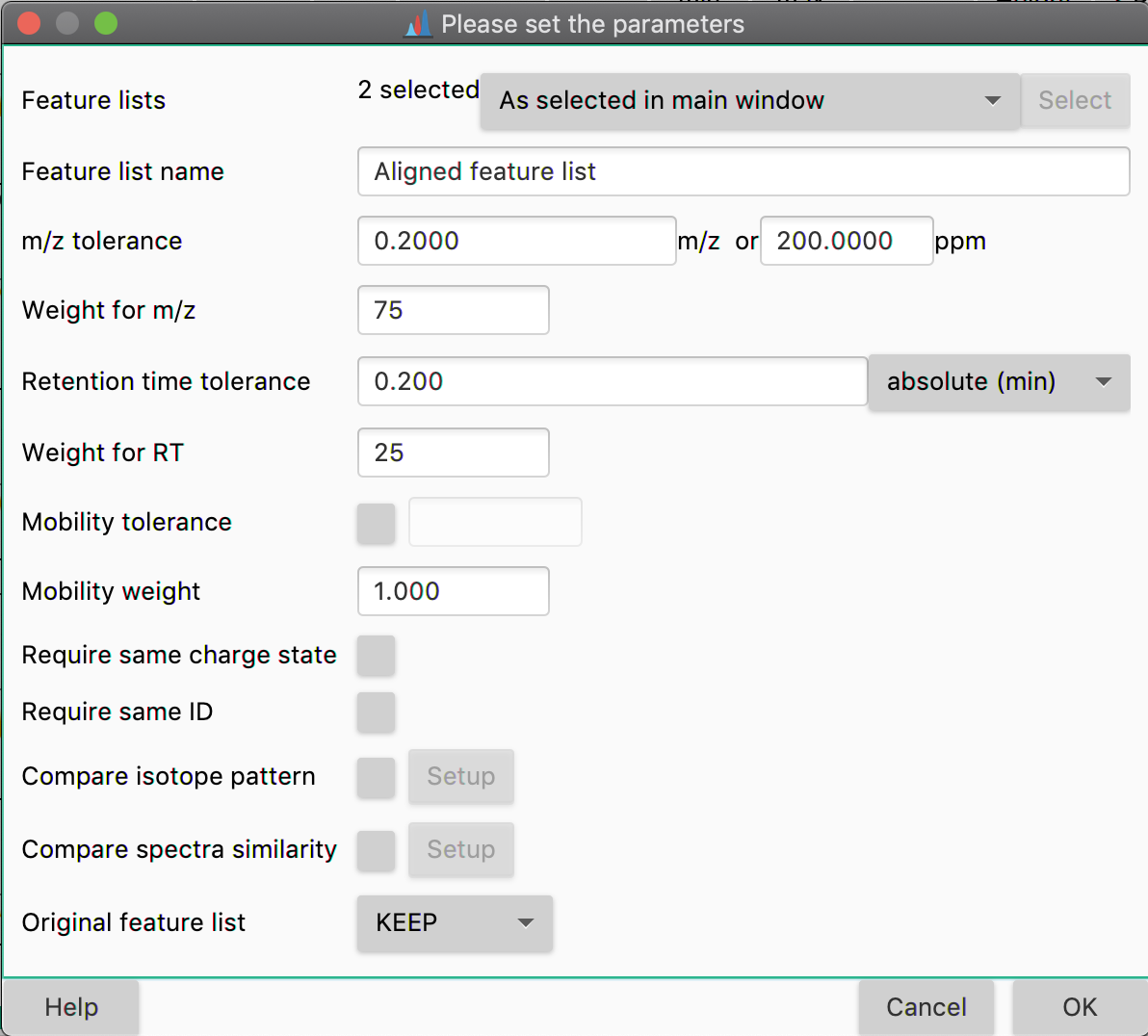
OKを押すことで実行します。
新しく出てきたAligned feature listをダブルクリックすると化合物リストが表示される。
クロマトグラムが2つあるデータは2つのデータに共通する化合物であり、1つのみのデータはどちらか一方にしかない可能性のある化合物である。
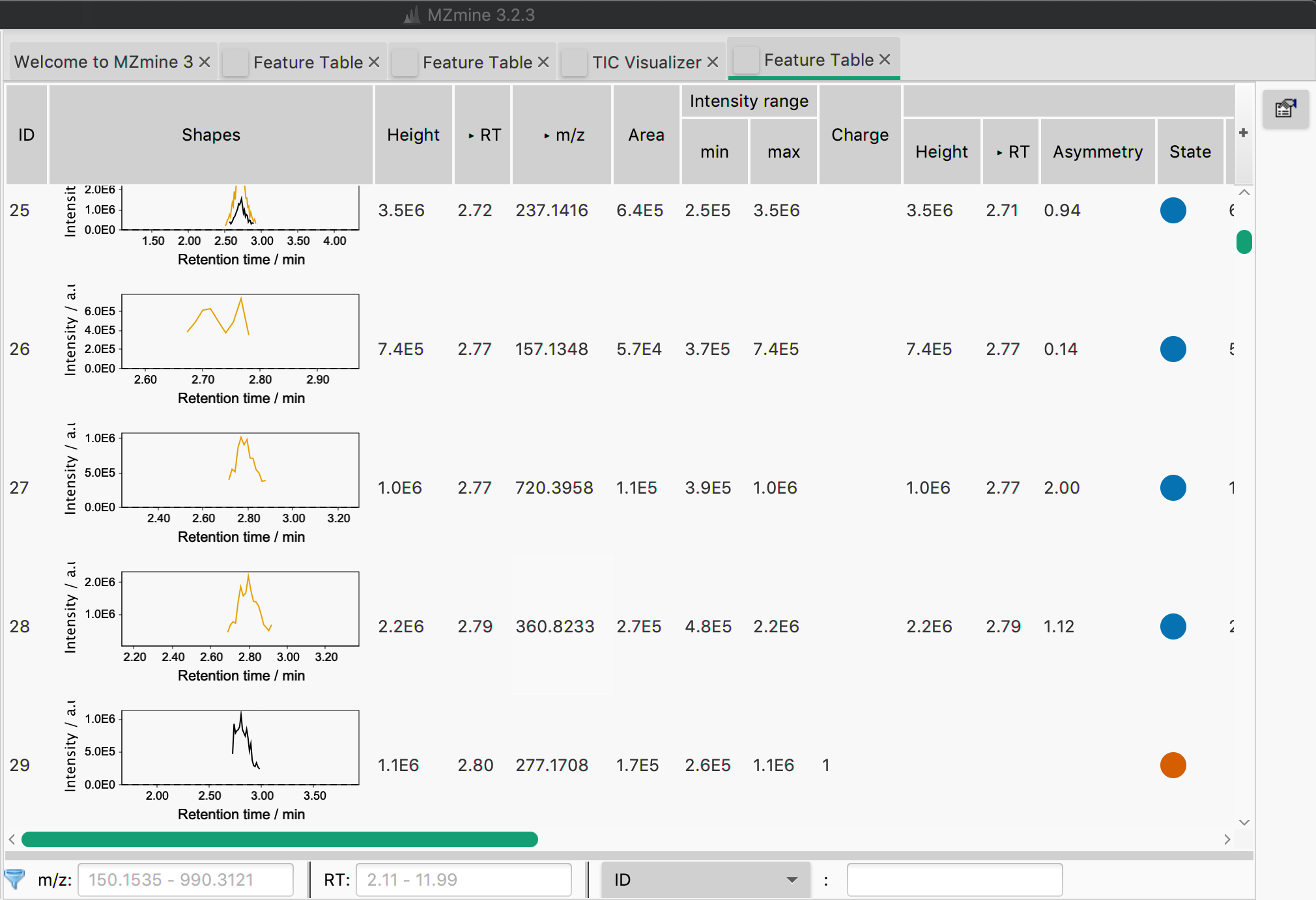
対象となる化合物を右クリックし、show > XIC (quick) もしくあ XIC (dailog) を用いて目的化合物の元データにおけるXICを確認することができる。
GNPS用にデータをエクスポート
GNPSはMSMSデータのネットワーク解析により、化合物の同定を試みる方法である。
Aligned feature listを選択する
Feature list methods > Export feature lists > GNPS -Feature-Based Molecular Networking- を実行する。
ファイル名を指定し、exportを実行する。
GNPSのサイトにアクセスし指定されたファイルをアップロードする (アカウントの作成が必要である)。