CryoSPARCは電子顕微鏡を利用した構造解析に用いられるソフトウェアである。
https://cryosparc.com/
商用利用は有料だがアカデミアは無償で利用できる。
ユーザーインターフェースは使いやすく、比較的簡単な操作で構造解析ができる。
1年以上利用してきたが、ここでは解析の一般的なスキームと分解能を向上させるために用いた方法論の備忘録をまとめる。
私はあくまで1ユーザーですし、経験も1年程度と浅いので、参考程度にしていただきたい。
解析は継続しているので、適宜更新していきたい。
より詳しい方でご意見がある場合はいただけるとありがたいです。
KEK、SBRCの資料やRelionの解説ページも参考にさせていただいております。
cryosparc seminarのYoutubeも参考にしています。
https://www.youtube.com/watch?v=eRPue0-Pkw4
https://www.youtube.com/watch?v=ehXqLnV_Jr4
https://www.youtube.com/watch?v=HXIipHORI2Y
起動に関して
terminalを起動し以下のコマンドでcryospacを起動する。
cryosparcm start
起動したPC上で、http://localhost:39000/をブラウザを用いて開くことで起動することができる。
ssh 接続で操作したい場合は以下のコマンドを実行し、パスを繋ぐことで、操作用のページを開くことができる。
ssh -l cryosparc -N -f -L localhost:39000:localhost:39000 ip_adress(PCNAME)
1. 一般的な構造解析スキーム
以下はとりあえず、解析を実施するときのイメージ図
一般的な方法論として記述しているが、template pickerは画像の品質がかなり高くないとうまく動かない印象を持つ。
そのため、自身がない場合はDeeppickerかTopazを用いた方が良いように思われる。

1.1. Import Movies
TitanもしくはTalosで測定した画像をeer形式で保存した形式を用いることが多いと思われる。
通常設定するパラメーターをいかに並べる。以降の表も基本的に同じである。
| 項目名 | 備考 | 通常設定する値の例 |
|---|---|---|
| Movies data path | eerの場所を入力する。*を用いてまとめてファイルを入力するとよい。*は2箇所入れても問題なく動作する。 | /movies/Supervisor_20230324_162049/Images-Disc1/GridSquare*/Data/*.eer |
| Gain reference path | eer movieのgain reference fileを指定。通常は測定時に作成したものを用いる。 | /movies/Supervisor_20230324_162049/20230320_103723_EER_GainReference.gain |
| Raw pixel size (A) | 画像の倍率に応じた値を入力する | 0.84など |
| Accelerating Voltage (kV) | 顕微鏡の電圧測定した機械に対応する値を入力 | 200 or 300 |
| Spherical Aberration (mm) | 顕微鏡固有の値。対応した顕微鏡に該当する値を入力する。 | 2.7 |
| Total exposure dose (e/A^2) | 測定に用いられた電子の量。 | 50 |
| EER Number of Fractions | 測定条件を調べ、適切に設定した方が良いという話もありますが、cryosparcの解説ページによるとほとんどの場合、40で問題ないようである。実際に変更したこともあるが、大きな違いは感じなかった。 | 40 |
| EER Upsampling Factor | 画像の分解能を上げることができる。1の場合は測定された画像のpixel sizeが半分の大きさになった画像が出力される。その分画像が重くなるため注意が必要である。元ものデータのクオリティが高くないと2にするメリットはないように思われる。なので基本的には1に設定した方が良い。 | 1 or 2 |
1.2. Patch Motion Correction
ImportしたMovieを解析にかける。
特にパラメーターを変更する必要はない。
1.3. Patch CTF Estimation
Patch Motion Correctionの出力を解析にかける。
特にパラメーターを変更する必要はない。
1.4. Manually Curate Exposures
Patch CTF Estimationの結果を出力する。
この工程はスキップしても良いが、解析にかける前に品質の良い画像をある程度選抜しておいた方が良いようである。
Average defocus
CTF fit resolution
Relative ice thickness
などいくつかのパラメーターを吟味し、ある程度優良な画像を選抜する。
最頻値から大きくずれている画像は除いた方が良いと考えています。
そのような画像の様子を目視で確認し、類似した数値をまとめ排除すると良いと考えています。
Manually Curate Exposuresの画面1

Manually Curate Exposuresの画面2
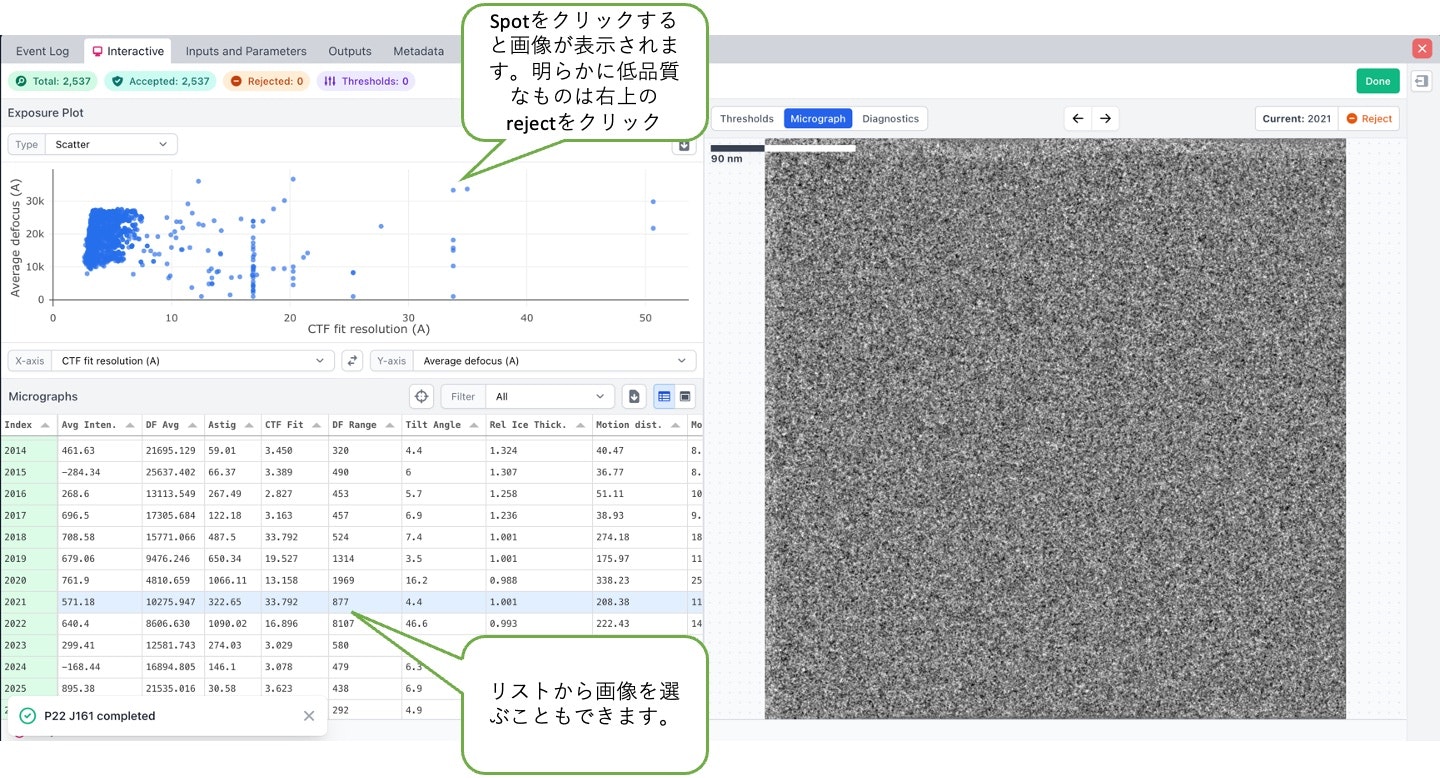
Manually Curate Exposuresの画面3
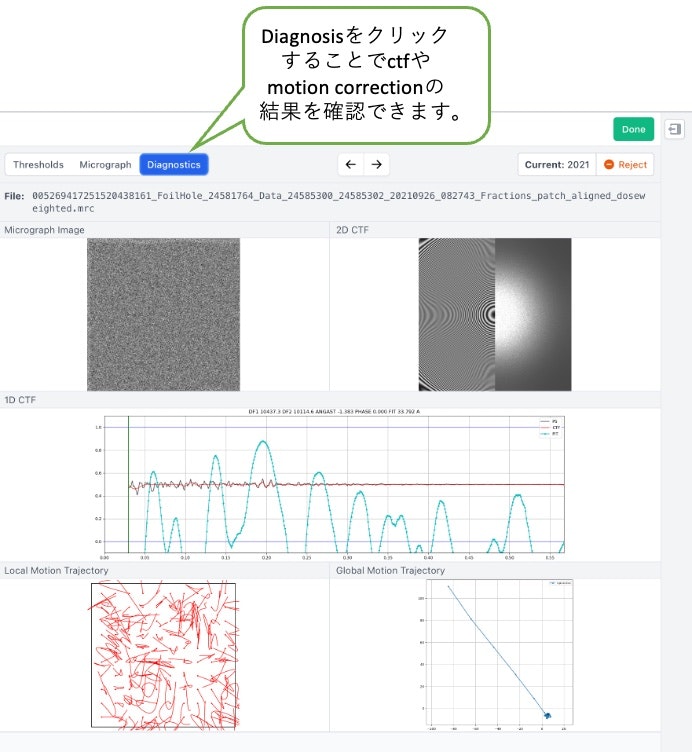
Manually Curate Exposuresの画面4

Manually Curate Exposuresの画面5 (品質の悪い画像のdiagnosis画面, CTFに白い傷のようなものがついている。)
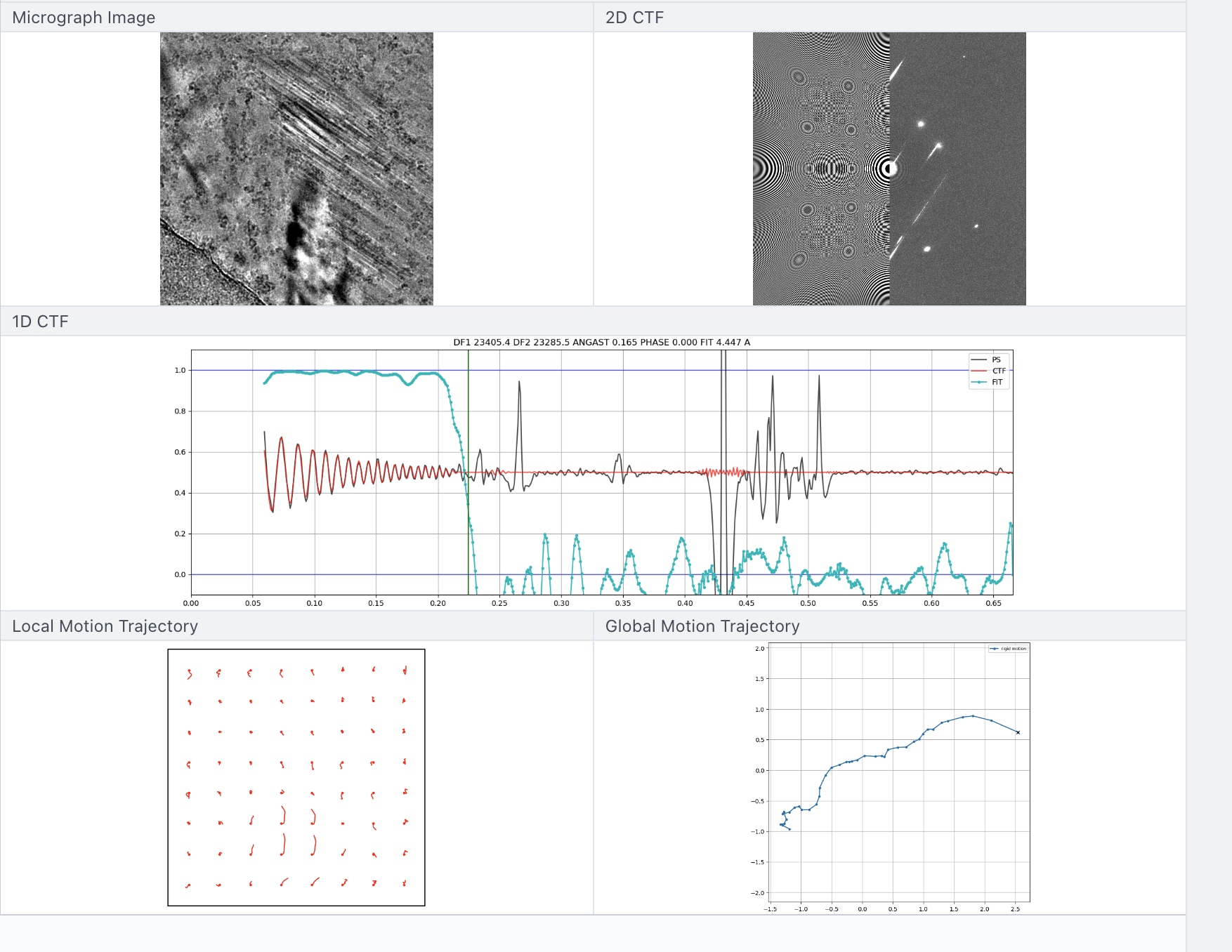
注:自身の経験談
CTF fit resolutionが悪いものは事前に除いた方が良い印象を持っています。
Relative ice thickness、Average defocusが極端なものは氷の状態が悪く、使えない画像が多い印象を持っています。
ただ、過度に除去すると粒子数が少なくなるデメリットが大きくなる場合もあるようです。
バランスが難しいと感じています。
1.4. Template pickのためのTemplateを作成
particleをpickするために通常は少数の画像からまずテンプレートを作成する。
ただ、particleによってはいきなりBlob Pickerを用いて全ての画像を処理しても問題ない。
1.4.1. Blob Picker
まず、template非依存のpickを行う。Manually Curate Exposuresを用いて選択した画像をもとに処理を行う。
| 項目名 | 備考 | 通常設定する値の例 |
|---|---|---|
| Min Particle diameter (A) | 予想される粒子サイズに応じて設定 | 200 |
| Max Particle diameter (A) | 予想される粒子サイズに応じて設定 | 250 |
| Number of mics to process | 実際に処理する画像の枚数。画像の品質にもよるが100から500でも良いと思われる。 | 500 |
1.4.2. Extract From Micrographs
Blob Pickerの結果を用いて画像を抽出する。
| 項目名 | 備考 | 通常設定する値の例 |
|---|---|---|
| Extraction box size (pix) | 予想される粒子サイズに応じて設定。コンピューターが処理しやすいpixelサイズがあるようなので、それを選んで指定する。 | 256, 320, 360, 400 |
| Fourier crop to box size (pix) | 画像を圧縮することができる。初期の解析では圧縮しても良い。数が多い場合は圧縮すると解析が早くなる。未入力な場合は実施しない。 | 62, 128, 80, 160, 180, 200, 未入力 |
box sizeの選択には以下のページを参考にしています。
https://blake.bcm.edu/emanwiki/EMAN2/BoxSize
Cryosparc guideのページ
https://guide.cryosparc.com/processing-data/tutorials-and-case-studies/performance-metrics
1.4.3. 2D Classification
抽出した粒子を用いて二次元分類を行う。
1.4.4. Select 2D Classes
2D Classificationで得られた画像をもとに良質な画像を選抜する。
十分良質な画像が得られた場合は選抜された画像を用いてtemplate pickを行う。
一方、良質な画像が得られない場合は比較的良質な画像を選択し、再度2D Classificationにかける。
1.4.5. 立体モデルを用いたtemplate作成
二次元分類の結果をtemplateとして用いても良いが、ある程度粒子数があるなら、以下の手順で3次元モデルをもとにしたtemplateを作成しても良い。
- Ab-Initio Reconstruction
- Homogeneous Refinement
- Create Templates
1, 2 に関しては詳しくは後述する。
Create TemplatesではNumber of equally-spaced templates to generateの数字を少し下げておいた方が良い。デフォルトは50だが20程度でも十分機能する場合が多いように思う。
1.5. Template picker
Select 2D ClassesもしくはCreate Templatesの出力とManually Curate Exposuresの出力を利用してtemplate pickを行う。あくまで、個人の感想ですが、Cryosparcのtemplate pickerはデータによってはほとんど、particleを選べていないように思う。Topaz Trainか後述する、Deep pickerかを用いた方が良いように思う。
| 項目名 | 備考 | 通常設定する値の例 |
|---|---|---|
| Particle diameter (A) | 予想されるparticleのサイズ。templateが存在してもこの値は必要である。 | 150, 200, 250, など |
| Lowpass filter to apply to templates (A) | templateにかけるフィルターの値。高いほどぼかしたものが利用される。デフォルトは20だがデフォルト設定だと経験上、ノイズを拾いすぎることが多いように思う。多少数字を下げておいた方が良いかもしれない。 | 5, 10, 15, 20 |
| Lowpass filter to apply to micrographs (A) | 電験画像にかけるフィルターの値。高いほどぼかしたものが利用される。デフォルトは20だがデフォルト設定だと経験上、ノイズを拾いすぎることが多いように思う。多少数字を下げておいた方が良いかもしれない。 | 5, 10, 15, 20 |
| Min. separation dist (diameters) | 粒子間の距離。特に変更する必要はない。particleが密な場合は数字を下げると良い。 | 0.5 |
1.6. Inspect Particle Picks
Template pickerの出力(MicrographsとAll particles)を用いて抽出する粒子の閾値を設定する。
画面右側で検出されている粒子の様子を確認しながら、画面左側のグラフのPower ScoreとNCC scoreの範囲を指定することである程度事前に粒子を排除しておく。
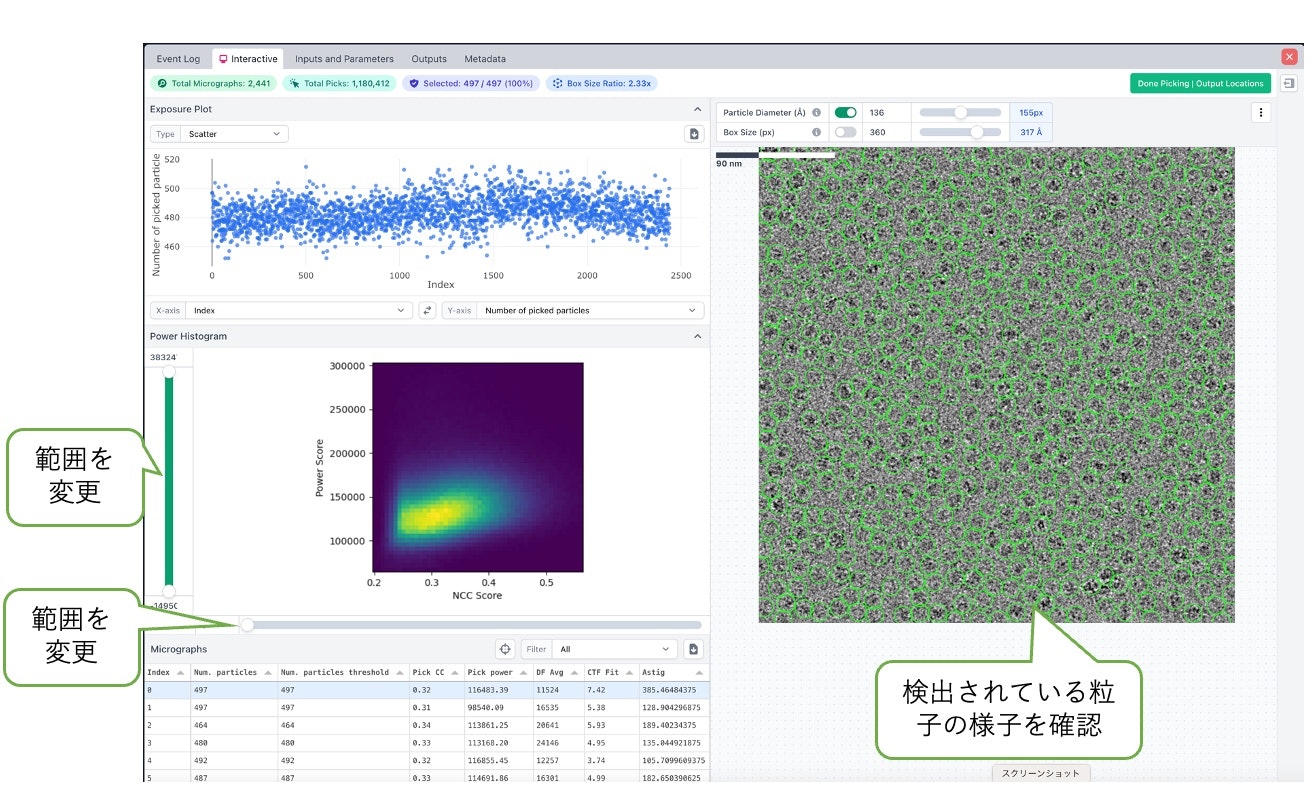
1.7. Extract From Micrographs(2回目)
次にモデル構築のための画像を抽出する。
Extraction box size (pix) を粒子の大きさに応じて設定する。粒子よりは大きめに設定した方が良い。
Fourier crop to box size (pix)は未入力でも良いが、particleサイズから見積もられる画像のサイズが大きすぎる場合あはExtraction box size (pix)の半分もしくは1/4程度を入力するとよい。
綺麗な粒子の選抜の過程である程度粒子が選抜できたら、高分解能の画像を再度抽出すると良い。
1.8. 良質な粒子の選抜
次に3次元マップを作成するために良質なparticleを選抜する。
1.8.1. 2D ClassificationとSelect 2D Classesを繰り返す。
最も基本的な方法である。これらを繰り返すことで良質な画像を選抜する。
以下のパラメーターは分類時に検討すると良いと思われる。
| 項目名 | 備考 | 通常設定する値の例 |
|---|---|---|
| Number of 2D classes | 出力するクラスの数。通常は50だが必要に応じて数字を大きくしてもいいように思われる。 | 150, 200, 250, など |
| Initial classification uncertainty factor | 分類を確定するまでにかける時間の基準。大きいほど、確定させるまでのサイクル数が多い。10まで増加させても良いようである。 | 2から10 |
| Circular mask diameter (A) | マスクサイズ。粒子の大きさに応じて変更する。20ごとにいくつか振って検討すると良い場合がある。 | 160, 180, 200, 220, 260など |
| Batchsize per class | 分類を繰り返す過程でのサイクルあたりに用いられるクラスあたりのparticleの数。デフォルトは100だが200まで増やすことがきる。 | 100, 200 |
1.8.2. Ab-Initio Reconstructionによる選抜
cryosparcにおいては3D モデル構築のプロセスでもある程度良質な粒子を選抜できるようである。
詳しくは事項にも記載。
1.9. 3Dマップ構築
1.9.1. Ab-Initio Reconstruction
初期3次元モデルを構築する。良質な粒子の選抜過程での最後の出力をin putとする。
| 項目名 | 備考 | 通常設定する値の例 |
|---|---|---|
| Number of Ab-Initio classes | 出力するクラスの数。デフォルトは1である。特に問題がなければ1で良い。複数のコンフォマーが予想される時やゴミとなる粒子が残っている可能性が高い場合は2~4を選択する。 | 1から4 |
| Maximum resolution | どの分解能までモデル構築を試みるか。 | デフォルトは12。ただし、小さいタンパク質など難しいものに対しては5程度に設定しても良いようである。 |
| Initial resolution | モデル構築初期の分解能 | デフォルトは35。ただし、小さいタンパク質など難しいものに対してはより小さい値に設定した方が良いらしい。Initial resolutionよりは大きい値で20-7の範囲で選んでも良さそう。 |
| Initial minibatch size | 分子量の小さいタンパク質や対称性の高いタンパク質に対しては大きくした方が良いようである。 | デフォルトは90, 1000程度までは少なくとも大きくできる。自動設定にすると小さいタンパク質で300に設定されるようである。 |
| Final minibatch size | 分子量の小さいタンパク質に対しては大きくした方が良いようである。 | 2000程度まで少なくとも大きくできる。自動設定にすると小さいタンパク質で2000に設定されるようである。 |
| Class similarity | 出力されるクラス間の類似度。デフォルトは0.1。ゴミを除きたい場合はここの数値を0に設定し、Number of Ab-Initio classesを2以上にするという方法論があるようである。 | 0から1 |
| Symmetry | デフォルトはC1。マップの対称性を指定する。マップ構築時に対称性を指定することもできるようだが、作成者によるとAb-Initioの段階で対称性を指定することは推奨されず、Refinementの過程で指定するのが良いようで有る。 | 基本C1, C2, D2など |
1.9.2. Homogeneous Refinement
3D modelのデータをさらに高分解能にする。
Ab-Initio Reconstructionで出力されたVolumeとparticleを用いる。
色々パラメーターがあるがとりあえず、Symmetry (C1, C2, D2 etc) のみをしてすれば良いように思われる。
| 項目名 | 備考 | 通常設定する値の例 |
|---|---|---|
| Symmetry | 対象性が破ることがわかっているなら対称性を入力する。そうでなければC1で良い。 | C1, C2, D2 etc |
| Initial lowpass resolution (A) | 解析を開始する初期の分解能。デフォルトは30。 インプットがrefinementのサンプルなら、インプットよりも少し悪い分解能を設定すれば良さそう。ただ、小さくしすぎるとあまり良くなさそう (特にAb-initioの直後)。大きさの小さいタンパク質の場合も数字を下げた方が良さそう (15, 20など)。 | 30, 20, 15, 5, 4, etc |
| Minimize over per-particle scale | デフォルトはオフだがオンにした方がよさそうである。 | on, off |
1.9.3. Non-uniform Refinement
Homogeneous Refinementで出力されたAll particles, Refined Volume, Maskを用いる。
色々パラメーターがあるがとりあえず、Symmetry (C1, C2, D2 etc) のみをしてすれば良いように思われる。
データの品質が十分良ければここまでの解析である程度良い分解能が出るはずである。
mapは2種類出力される。mapとmap sharpである。mapが最初に出力されるもので、sharpはB-factorに基づいてmapを改良したものである。
改良されたmapの方が基本的にモデル構築に適しているようであるが、分解能やデータによっては用いるべきではないようである。mapとmap-sharp双方を取得し、よく確認した上で、どちらをモデル構築に用いるか考える必要がある。
詳しくは以下のページを見ていただけると良いかと思います。
1.9.4. Local resolution estimation
Non-uniform Refinementの出力を用いて局所分解能を推定する。
2. 分解能をさらに改善するために考えられる対策
Cryosparcのディスカッションパネルで見かけた方法などから自信で試した経験があるものとその結果の評価をまとめています。現在工事中です。
cryosparcの掲示板でもいくつか分解能改善方法について議論されています。
https://discuss.cryosparc.com/t/strategies-for-high-resolution-refinement/5587
2.1. Homogeneous Refinement、Non-uniform Refinementの設定変更
全てのデータではないが、「Minimize over per-particle scale」をオンにした場合、分解能の向上が見られる場合があるようである。
また、「Minimize over per-particle scale」をオンにした状態で「Optimize per-group CTF params」をオンにすることでさらに若干分解能が改善する場合もった。「Optimize per-group CTF params」の影響はHomogeneous Refinementの際により効果的な印象がある。
「Minimize over per-particle scale」と「Optimize per-group CTF params」がをオンの「Homogeneous Refinement」と「Minimize over per-particle scale」がオンの「Non-uniform Refinement」のを繰り返す操作で若干分解能が改善した。
2.1.1. 他のパラメーターを変更してみた経験(あくまで、一例です。)
2.1.1.1. Fit Spherical Aberration、Fit Tetrafoil、Fit Anisotropic Magをオンにした場合
一時的に分解能が向上した場合もあるが、mapの品質は低下しているように見えた。
2.2. Motion Coorectionの利用
Local motion CoorectionなどによりMotion Correctionをやり直すことで分解能が向上することがあるようです。
また、v.4.4からreference based motion correctionが実装されました。Relionにおける、Bayesian polishingに類似した機能のようです。計算時間はかなりかかりますが、分解能の向上は見込める方法論になります。
cryosparcにおける、reference based motion correctionの解説ページ
https://guide.cryosparc.com/processing-data/all-job-types-in-cryosparc/motion-correction/job-reference-based-motion-correction-beta
2.2.1. Local Motion Correction
Local Motion Correctionを行うと分解能が伸びたという報告があります。以下は私が実施した方法です。
2.2.1.1. Local Motion Correction
Curate ExposureのMicrographとNon-uniform Refinementのparticlesをinputとして解析を実施しました。
基本的にパラメーターはデフォルトで行った。
Extraction box size (pix)のみ指定した(上述のExtract From Micrographsと同じ値を用いた。)。
2.2.1.2. Ab-Initio Reconstruction
そのまま、解析しようとしたが、なぜかノイズがかなり大きくなっていた。そこで、Ab-Initio Reconstructionのパラメーターを変更して実施し、質の悪い粒子を除くこととした。
Number of Ab-Initio classes: 2に設定し、実行した結果、大部分の良質なクラスと少量の質の悪いクラスに分離した。
2.2.1.3. Homogeneous Refinement、Non-uniform Refinementの実施
「Optimize per-group CTF params」をオンにしこれらを連続して実施すると改善する場合があるようである。
2.2.2. reference based motion correction
実際に0.2Å程度自分の例では分解能が向上しました。
2.2.2.1. Homogenous refinementの実施
Minimize over per-particle scaleをonにして一度refinementを実施します。
2.2.2.2. reference based motion correction
インプットに必要な情報はparticle, refined volume, mask, micrographsです。
特にパラメーターを変更する必要はないようです。
box sizeを指定する項目がなく、不安に思いましたが、in putしたparticlesと同じbox sizeのparticleが出力されてくるようです。
出力されたparticlesを用いて再度refinementを行うと分解能が向上しました。
2.3. Junk particleの除去
Junk particleを除く方法は以下の掲示板で議論されていました。
いくつか分類してまとめておきます。
2.3.1. Ab-inito reconstruction
Number of Ab-Initio classesを2以上、Class similarity
を0に設定し、Ab-Initio Reconstructionを行う。
2.3.2. 優良なクラスとゴミクラスを用いてHeterogeneous Refinementを行う
2.3.3. Ab-initio reconstructionとHeterogeneous Refinementを繰り返す。
下記ページで、 議論されている方法である。
Number of Ab-Initio classesを5にしてAb-initio reconstructionを実施し、出力されたデータを用いてHeterogeneous Refinementを実施する。
Heterogeneous RefinementにはAb-initio reconstructionにあるAll particlesと出力されたVolumeを全て入力値とする。
良いクラスを選抜する。
良いクラスを用いてNumber of Ab-Initio classesを5にしてAb-initio reconstructionを実施し、同様の作業を行う。
2.4. Box sizeの検討
Particle extractionのbox sizeを変更した場合の例
Defocusにもよるようだが、box sizeはある程度大きい方が良い。
タンパク質X, 粒子の大きさは200×50×50Å程度, 0.84 Å/pixelの画像、particle数: 51852の場合
それぞれに対してparticleの再抽出後、
- Ab-initio reconstruction
- Homogeneous refinement (C1; Minimize over per-particle scale, on)
- Non-uniform refinement (C1; Minimize over per-particle scale, on; Optimize per-group CTF params, on)
を実施
box sizeを大きくした際に、particleの数が少し減少してしまった。ある程度、改善が見られた。
| 3後の分解能 | Box size (pixcel) | Box size (Å) | particle |
|---|---|---|---|
| 3.55 | 360 | 302.4 | 51852 |
| 3.35 | 400 | 336 | 50871 |
| 3.33 | 416 | 349.44 | 50515 |
| 3.36 | 440 | 369.6 | 49937 |
| 3.40 | 480 | 403.2 | 48784 |
2.5. 3D classificationの利用
Refinment結果を3D classificationに投げることで、分類を行う。
ParticlesとSolvent maskの入力は必須。initial volumeの入力は不要。入力する場合は複数入力する必要がある。
| パラメーター名 | 解説 | 数値例 (default) |
|---|---|---|
| O-EM learning rate init | (調査中) | 0.75 (0.4) |
| O-EM learning rate half-life (iters) | (調査中) | 100(50) |
| RMS density change convergence check | (調査中) | on, (off) |
| Per-particle scale | noneは小さいparticleや奇妙なparticleの時は設定するとよいと書いてある。 | none, in put, (optimum) |
| Force hard classification | 小さいターゲットの時はonにすると良いと書いてある。 | on, (off) |
| Class similarity | 出力される構造同士の類似度を指定することができる。auto-tune initial class similarityという項目も存在する。 | 0.25(0.5) |
参考質疑応答
https://discuss.cryosparc.com/t/3d-classification-all-classes-are-the-same/8296
2.5.1 Focused 3D classificationの利用
特定の領域のフォーカスして3d classificationを実施する。
一部のマイナーなコンフォメーションを見つけるために役に立つ可能性もある。
実際、私も実施し、そのままでは観測できなかった部分構造が観測できるようになった経験はある。
Focused maskを作成し、ParticlesとSolvent maskに加え、入力することで、特定の場所にフォーカスしてクラス分けすることができる。
2.5.1.1 Focused maskの作成方法
大きく分けて2つの方法が考えられる。
- Solvent maskをダウンロードし、不要な箇所を削除する。
- mapを元に、pdbモデルを作成し、フォーカスしたい領域近傍以外を削除する。得られたpdbを元にChimeraXを利用してmapを作成する。
3. その他
3.1 Deep pickerを使ってみた
ある程度の分解能で解けた構造をもとにDeep pickerを実施してみました。
実際、この方法が正しいかわかりませんが、これにより得られたparticleとそれ以前に得られたpartcleを合わせて構造解析をさらに行った結果、若干分解能が改善しました。
Non-uniform refinementで出力されて構造を入力にしました。
3.1.1. Deep Picker trainの実施
Deep Pickerで何らかの理由で取り漏らしているparticleを追加することで分解能を改善できないか検討した。
Microgpraphsには最も上流のCurate exposureの出力を用いた。
Particlesは最後に実施したNon-uniform refinementのparticleを用いた。
Modelは不要である。
何も考えずに実施したら大体エラーで停止しました。
おそらくGPUのメモリが原因だと考えています。
なので、Number of micrographs to processを
100, 200, 300, 500, 800, 1000といくつか変更し、入力しました。
Particle diameter (A)もparticleの最も大きい辺のサイズを入力しました。
学習が適切に終了する中で最も大きいNumber of micrographs to processを用いた出力を用いて次の解析を行いました。
3.1.2. Deep Picker Inferenceの実施
Microgpraphsには最も上流のCurate exposureの出力を用いた。
Modelには3.1.1.の出力を用いた。
これによりある程度のparticleが得られた。
3.1.3. 以降に実施した操作の例
- Extract from micrograph
- Ab-initio reconstruction (Number of Ab-Initio classes: 2, Class similarity: 0)
- Ab-initio reconstructionをもう一度繰り返し、junkなparticleを除いた
- Remove Duplicate particleを実施し、trainに使ったparticleと新しく得たparticleの重複を除いた。
- Ab-initio reconstructionを同様の設定で何度か繰り返し、junkなparticleを除いた
- Homogeneous refinementとNon-uniform refinementの実施
最終的にはparticleの数が3割増していどになり、分解能は0.1~0.2程度上昇した。
ただ、この操作の後、Homogeneous refinementをOptimize per-group CTF paramsをオンで実施しようとするとエラーが出るようになった。
原因は不明である。
3.2 3D Valiabilityを使ってみた
- 最後に実施したNU-refinementの出力のparticleをMaskをin putとして用いる。
- Filter resolution (A)の入力は必須である。
- 終了後、3D Variability Displayに出力データを投げ、解析を行う。
- 出力されたVolumeをダウンロードする。
- ChimeraXを用いてvolumeを可視化する。
ChimeraXに以下のようなコマンドを入力することでファイルの動きを可視化する。
open folder_path/cryosparc_P20_J1328_component_001/J1328_component_001_frame_*.mrc vseries true
タイル表示したい場合は以下の手順を踏むと良い。
open folder_path/cryosparc_P20_J1328_component_001/J1328_component_001_frame_*.mrc
#タイル表示する。
tile
#間隔を調整する。間隔を広げる場合は0.6~0.8、広げる場合は1.2~1.5を設定するといい。
tile spacingFactor 0.8
#mapの表示具合を調整する。
# 細かくみたい場合はstep 1に全てのmapを変更する。
volume step 1
#比較したい閾値を設定する。
volume level 0.015
4 Relionへのデータ移行
relionでしか使えないツールを利用する場合、Relionにデータを移行する必要がでてくる。
pyemツールを用いてcryosparcとrelion間のデータのやり取りができるようである。
この方法で3d classificationなどは利用できるがBayesian polishingなどは利用できない。
詳しくは以下のページを参照してほしい。
https://github.com/asarnow/pyem/wiki/Export-from-cryoSPARC-v2#particles-extracted-outside-cryosparc
4.1 cryosparcの解析結果をrelion用のstarファイルに変換し、relionにインポートする。
cryosparcのAb-initio reconstruction, homogeneous refinementを用いて得られたmapをrelion用に出力し、3d classification without alignmentを実施することなどを想定している。
まず、マップ作成に用いられたExtract from micrographsのjobとhomogeneous refinementのjobのpathを確認する。
対象のjobをクリックし、DetailsのDirectoryを確認することで把握することができる。
二つのjobを用いて解析を行う。
#Extract From MicrographsのDirectoryの例
/run/media/cryosparc/folder/P20/J780
#homogeneous refinementのDirectoryの例
/run/media/cryosparc/folder/P20/J819
フォルダとファイルの準備
#relion用のDirectoryを作成する。
mkdir relion-project
cd relion-project
#particleのmrcファイルをリンクするためのフォルダを作成する。さらに作成したフォルダに移動する。
#J780はExtract from micrographsのjobに該当する。
mkdir -p J780/extract
cd J780/extract
次にmrcファイルへのリンクを貼ります。
relionはmrcを読み込めないため、mrcsとリンク名を変更します。
#particleのmrcファイルへのリンクを貼る。
ln -s /run/media/cryosparc/folder/P20/J780/extract/*.mrc -t .
rename .mrc .mrcs *.mrc
#元のフォルダに戻る。
cd ../..
ファイル数が多いとln -sやrenameにエラーが出て走らない。
必要に応じてシェルスクリプトを用いると良い。
以下のコマンドでrelion用のstarファイルを作る。
今回はhomogeneous refinementのDirectoryの情報を用いる。
csparc2star.py /run/media/cryosparc/folder/P20/J819/J819_003_particles.cs \
/run/media/cryosparc/folder/P20/J819/J819_passthrough_particles.cs \
from_csparc.star
sed -i 's/particles.mrc/particles.mrcs/g' from_csparc.star
次にrelionにインポートします。
Importをクリックする。
Movies/micsタブ設定
| 項目 | 設定値 |
|---|---|
| Import raw movies/micrographs? | no |
Othersタブの設定
| 項目 | 設定値 |
|---|---|
| Import other node types? | yes |
| Import file | from_csparc.star |
| Node type | Particles STAR file (.star) |
これによりrelionにデータをインポートすることができた。
4.2 Relionでno alignment Class 3Dを実行し、出力情報をcryospacに戻すまで。
4.2.1 no alignment Class 3Dの実行
上述の方法でimportしたファイルを元にrelionで3D classificationを実施する。
| タブ | 項目 | 設定値 |
|---|---|---|
| I/O | Input imgaes STAR file | 先ほどimportした項目を利用する。 |
| I/O | Reference map | 対応するcryosparc jobのマップのmrcファイルを用いる。 |
| Reference | Ref. map is on absolute greyscale? | Yes |
| Reference | Initial lo-pass filter (A) | 0 |
| Reference | Symmetry | C1など、構造に応じて適切に設定。 |
| CTF | Do CTF-correction? | Yes |
| CTF | Ignore CTFs until first peak? | Yes |
| Optimization | Numberof classes | 2 |
| Optimization | Regularisation parameter T | 4,8,16,32,64など、検討する |
| Optimization | Use fast substes | no |
| Optimization | Mask diameter | 構造に応じて適切に指定 |
| Optimization | Mask individual particles with zeros | Yes |
| Optimization | Limit resolutions E-step to (A) | -1 |
| Sampling | Perform image alignment | no |
4.2.2 Subset selection
これにより、前の操作で得られたものからクラスを選ぶ。
| タブ | 項目 | 設定値 |
|---|---|---|
| I/O | Select class from job | 先ほどのclass 3Dの結果を選ぶ |
どちらかのClassを選び、保存する。
それぞれのClassについて実行する。
4.2.3 Cryosparcへのデータの読み込み
subset selectionで出力さたparticles.starを多少修正しい、cryosparcにインポートする。
上述の手順でcryosparcからrelionに移動させたデータを再読み込みすることを前提にしています。
relion解析フォルダに移動し以下の操作を実行する。
#Job 番号は適切に修正
cd Select/job023
#particles.starファイルを複製する。
cp particles.star particles2.star
#particles2.starファイル内のmrcs形式の記述を全てmrcに戻す。cryosparc側ではmrcを持ちいているため。
sed -i 's/particles.mrcs/particles.mrc/g' particles2.star
Cryosparc側、Builder Job、Import Particle Stackを選ぶ。
Source micrographにExtract from micrographsのjobのimportに用いたmicrographを選択する(選択しなくても大丈夫なようである)。
Particle meta path: 上述のparticles2.starを選択する。
Particle data path: cryosparcのroot folderを選択する。プロジェクト番号のフォルダまで(例:/your_path/cryosparc/P20/)
上述のstarファイル中では画像のファイル名は以下のように登録されているようである(上述のようにcryosparcの由来のファイルを元にしていた場合)。cryosparcのextractparticlesは/J000/extract/フォルダに保存されているので上記の設定で適切に動作するようである。
J780/extract/000000047731243579427_FoilHole_22862148_Data_22953862_22953864_20230421_193752_EER_patch_aligned_doseweighted_particles.mrc
インポートしたprticlesとvolumeを用いてhomogeneous refinementを実施すれば良い。
4.3. PDBからマスクを作成する。
- mapファイルをダウンロードする。
- mapファイルをchimeraXで開く(#1となる想定)。
- モデルのpdbファイルを開く (#2となる想定)。
- pdbファイルをModelsをmodels windowで選択する。
- Right MouseタブのMove Modelをクリックし、右ドラッグで構造をmapの近くに移動させる。
- Right MouseタブのRotate Modelをクリックし、右ドラッグで構造とmapが重なるよう角度を調整する。
- 誤って動かしてしまわないようRight MouseタブのSelectを選んでおく。
- MaptタブのFitをクリックするとpdbがマップにハマるはずである。うまくはまらないようなら再度調整、もしくはマップをFlipさせる(今後のこともあるので、cryosparcのVolume tools, Flip handをonにして実行し、Flip後、refinementをやり直しておくと良いと思われる。)。
- 以下のコマンドを実行。出力されたmapをmrc形式で保存する。
- Import Volumeでcryosparcにimportする
a. Volume data pathにファイル名を設定する。
b. Type of volume being importedをmaskに設定する。 - Volume Toolsでmaskとして利用できるように加工する。
a. Static maskにimportしたVolumeをインポートする
b. Type of input volume, Type of output volumeをmaskに設定する。
c. Thresholdを設定する。ChimeraXでlevelを調整し、目的の範囲を覆うようなlevelを探す。これにより得られた値をここに入力する。~0.04あたりが良いと思われる。
d. Soft padding widthに数値を入力する。2-10程度で良いと思われる。解析に使うためにこの値の設定も必須の様である。
#pdbを元にマップを作成する。20は目的タンパク質に応じて設定するとよい。
#出力されたmapは#3になるはずである。
molmap #2 20
#補足です。以下のような表記にすれば部分構造を指定することも可能です。
molmap #3/A:777-965 20
#gridをインポートしたマップに合わせる。これを行わないと解像度などが元のデータと同じにならない。
vop resample #3 onGrid #1
注:cryosparcのVolume toolsの代わりに事前に以下のような操作をしておくこともできるが、Volume toolsの方が簡便である。
#scaleをさらに以下の操作で調整した方がいい。(この操作はcryosparcのSet volumes toolで代用した方が良いかもしれません。)
#全体的に数字が小さすぎるのでscaleを大きくした方が良い。#<sample>の数字は適宜変更
#shiftやthresholdの値もmapを見ながら調整すると良さそう。
vop scale #3 shift 0.08
#全体的に数字が小さすぎるのでscaleを大きくした方が良い。
vop threshold #4 maximum 0.17 setMaximum 1
参考ページ(chimeraコマンド)
5 cryosparcの取り扱いに関して
5.1 Cryosparcのscratchのデータを削減したい場合。
Scratchの寿命を変更し、cryosparcを再起動すると良い。
参考ページ
#viでcryosparc_master内にある、config.shを開く。cryosparc_masterの所在はインストールした場所次第。
vi cryosparc_master/config.sh
#以下の行を追記する。数字は目的に合わせて調整。
export CRYOSPARC_SSD_CACHE_LIFETIME_DAYS=15
#cryosparcの再起動
cryosparcm restart
古いScratchのファイルを自動で削除するようになります。